 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1pgt: CRYSTAL STRUCTURE OF HUMAN GLUTATHIONE S-TRANSFERASE P1-1[V104] C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1pgt | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN GLUTATHIONE S-TRANSFERASE P1-1[V104] COMPLEXED WITH S-HEXYLGLUTATHIONE | ||||||
 Components Components | GLUTATHIONE S-TRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE / PI CLASS / HGSTP1-1[V104] / DETOXIFICATION | ||||||
| Function / homology |  Function and homology information Function and homology informationnitric oxide storage / S-nitrosoglutathione binding / cellular response to cell-matrix adhesion / TRAF2-GSTP1 complex / negative regulation of smooth muscle cell chemotaxis / negative regulation of leukocyte proliferation / dinitrosyl-iron complex binding / common myeloid progenitor cell proliferation / hepoxilin biosynthetic process / glutathione derivative biosynthetic process ...nitric oxide storage / S-nitrosoglutathione binding / cellular response to cell-matrix adhesion / TRAF2-GSTP1 complex / negative regulation of smooth muscle cell chemotaxis / negative regulation of leukocyte proliferation / dinitrosyl-iron complex binding / common myeloid progenitor cell proliferation / hepoxilin biosynthetic process / glutathione derivative biosynthetic process / response to L-ascorbic acid / linoleic acid metabolic process / Glutathione conjugation / negative regulation of monocyte chemotactic protein-1 production / nitric oxide binding / JUN kinase binding / oligodendrocyte development / glutathione peroxidase activity / Paracetamol ADME / negative regulation of stress-activated MAPK cascade / negative regulation of JNK cascade / prostaglandin metabolic process / cellular response to glucocorticoid stimulus / regulation of stress-activated MAPK cascade / negative regulation of interleukin-1 beta production / Detoxification of Reactive Oxygen Species / negative regulation of acute inflammatory response / glutathione transferase / animal organ regeneration / negative regulation of ferroptosis / response to amino acid / negative regulation of vascular associated smooth muscle cell proliferation / glutathione transferase activity / negative regulation of tumor necrosis factor production / negative regulation of fibroblast proliferation / protein serine/threonine kinase inhibitor activity / negative regulation of tumor necrosis factor-mediated signaling pathway / regulation of ERK1 and ERK2 cascade / toxic substance binding / negative regulation of MAPK cascade / positive regulation of superoxide anion generation / xenobiotic metabolic process / cellular response to epidermal growth factor stimulus / negative regulation of extrinsic apoptotic signaling pathway / fatty acid binding / negative regulation of canonical NF-kappaB signal transduction / glutathione metabolic process / response to reactive oxygen species / central nervous system development / negative regulation of ERK1 and ERK2 cascade / cellular response to insulin stimulus / response to estradiol / cellular response to lipopolysaccharide / secretory granule lumen / vesicle / ficolin-1-rich granule lumen / response to ethanol / Neutrophil degranulation / negative regulation of apoptotic process / negative regulation of transcription by RNA polymerase II / mitochondrion / : / extracellular exosome / extracellular region / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.8 Å | ||||||
 Authors Authors | Ji, X. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1997 Title: Structure and function of the xenobiotic substrate-binding site and location of a potential non-substrate-binding site in a class pi glutathione S-transferase. Authors: Ji, X. / Tordova, M. / O'Donnell, R. / Parsons, J.F. / Hayden, J.B. / Gilliland, G.L. / Zimniak, P. #1: Journal: Biochemistry / Year: 1994Title: Structure and Function of the Xenobiotic Substrate Binding Site of a Glutathione S-Transferase as Revealed by X-Ray Crystallographic Analysis of Product Complexes with the Diastereomers of 9- ...Title: Structure and Function of the Xenobiotic Substrate Binding Site of a Glutathione S-Transferase as Revealed by X-Ray Crystallographic Analysis of Product Complexes with the Diastereomers of 9-(S-Glutathionyl)-10-Hydroxy-9,10-Dihydrophenanthrene Authors: Ji, X. / Johnson, W.W. / Sesay, M.A. / Dickert, L. / Prasad, S.M. / Ammon, H.L. / Armstrong, R.N. / Gilliland, G.L. #2: Journal: Eur.J.Biochem. / Year: 1994Title: Naturally Occurring Human Glutathione S-Transferase Gstp1-1 Isoforms with Isoleucine and Valine in Position 104 Differ in Enzymic Properties Authors: Zimniak, P. / Nanduri, B. / Pikula, S. / Bandorowicz-Pikula, J. / Singhal, S.S. / Srivastava, S.K. / Awasthi, S. / Awasthi, Y.C. #3: Journal: J.Mol.Biol. / Year: 1992Title: Three-Dimensional Structure of Class Pi Glutathione S-Transferase from Human Placenta in Complex with S-Hexylglutathione at 2.8 A Resolution Authors: Reinemer, P. / Dirr, H.W. / Ladenstein, R. / Huber, R. / Lo Bello, M. / Federici, G. / Parker, M.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1pgt.cif.gz | 103.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1pgt.ent.gz | 78.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1pgt.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/pg/1pgtftp://data.pdbj.org/pub/pdb/validation_reports/pg/1pgt | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2pgtC  1gssS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.94607, -0.108441, 0.305274), Vector: |
-Components
| #1: Protein | Mass: 23363.742 Da / Num. of mol.: 2 / Mutation: VAL 104 VARIANT Source method: isolated from a genetically manipulated source Details: HGSTP1-1[V104] AND HGSTP1-1[I104] ARE NATURALLY OCCURRING VARIANTS OF HGSTP1-1 OBTAINED BY SITE-DIRECTED MUTAGENESIS Source: (gene. exp.) Homo sapiens (human) / Cell line: 293 / Cellular location: CYTOPLASM / Gene: GTP_HUMAN / Organ: PLACENTA / Plasmid: BL21 / Production host:  #2: Chemical | 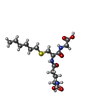  Mass: 392.491 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H30N3O6S Mass: 392.491 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H30N3O6S#3: Chemical |   Mass: 238.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM Mass: 238.305 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C8H18N2O4S / Comment: pH buffer*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 326 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 326 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 49 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion, hanging drop / pH: 6.5 Details: CRYSTALS WERE GROWN IN HANGING DROPS WHICH INITIALLY CONSISTED OF 5.9 MG/ML PROTEIN IN 0.1 M HEPES BUFFER (PH 6.5) CONTAINING 8.3 MM S-HEXYLGLUTATHIONE AND 1.0 M BUFFERED (PH 6.5) AMMONIUM ...Details: CRYSTALS WERE GROWN IN HANGING DROPS WHICH INITIALLY CONSISTED OF 5.9 MG/ML PROTEIN IN 0.1 M HEPES BUFFER (PH 6.5) CONTAINING 8.3 MM S-HEXYLGLUTATHIONE AND 1.0 M BUFFERED (PH 6.5) AMMONIUM SULFATE. THE DROPS WERE EQUILIBRATED AT 293 K AGAINST WELL SOLUTION CONTAINING BETWEEN 1.9 - 2.0 M AMMONIUM SULFATE IN 0.1 M HEPES BUFFER (PH 6.5)., vapor diffusion - hanging drop | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR571 / Wavelength: 1.5418 |
| Detector | Type: MACSCIENCE / Detector: IMAGE PLATE / Date: Mar 1, 1996 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→30 Å / Num. obs: 43132 / % possible obs: 96 % / Observed criterion σ(I): 0 / Redundancy: 4.2 % / Biso Wilson estimate: 24.48 Å2 / Rmerge(I) obs: 0.057 / Net I/σ(I): 13.92 |
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 1.41 % / Rmerge(I) obs: 0.303 / Mean I/σ(I) obs: 1.81 / % possible all: 86.9 |
| Reflection shell | *PLUS % possible obs: 86.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1GSS Resolution: 1.8→6 Å / Cross valid method: THROUGHOUT / σ(F): 2 Details: X-PLOR IS USED AT EARLY STAGE OF REFINEMENT. GPRLSA IS A MODIFIED VERSION OF PROLSQ BY FUREY, WANG AND SAX (J. APPL. CRYSTALLOGR.,1982,15,160-166). CROSS-VALIDATION INCLUDES GEOMETRY CHECK ...Details: X-PLOR IS USED AT EARLY STAGE OF REFINEMENT. GPRLSA IS A MODIFIED VERSION OF PROLSQ BY FUREY, WANG AND SAX (J. APPL. CRYSTALLOGR.,1982,15,160-166). CROSS-VALIDATION INCLUDES GEOMETRY CHECK AND R FACTOR CALCULATION FOR ALL DATA.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.94 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→6 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: GPRLSA / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(I): 1 / Rfactor obs: 0.182 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
