Entry Database : PDB / ID : 1px7Title A folding mutant of human class pi glutathione transferase, created by mutating aspartate 153 of the wild-type protein to glutamate Glutathione S-transferase P Keywords / / / / Function / homology Function Domain/homology Component
/ / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / / Biological species Homo sapiens (human)Method / / Resolution : 2.03 Å Authors Kong, G.K.-W. / Polekhina, G. / McKinstry, W.J. / Parker, M.W. / Dragani, B. / Aceto, A. / Paludi, D. / Principe, D.R. / Mannervik, B. / Stenberg, G. Journal : To be Published Title : The multi-functional role of a highly conserved aspartic acid residue in glutathione transferase P1-1Authors : Kong, G.K.-W. / Polekhina, G. / McKinstry, W.J. / Parker, M.W. / Dragani, B. / Aceto, A. / Paludi, D. / Principe, D.R. / Mannervik, B. / Stenberg, G. History Deposition Jul 2, 2003 Deposition site / Processing site Revision 1.0 Jul 22, 2003 Provider / Type Revision 1.1 Apr 29, 2008 Group Revision 1.2 Jul 13, 2011 Group Revision 1.3 Oct 11, 2017 Group / Category / Item / _software.nameRevision 1.4 Nov 10, 2021 Group / Derived calculationsCategory database_2 / pdbx_struct_conn_angle ... database_2 / pdbx_struct_conn_angle / struct_conn / struct_ref_seq_dif / struct_site Item _database_2.pdbx_DOI / _database_2.pdbx_database_accession ... _database_2.pdbx_DOI / _database_2.pdbx_database_accession / _pdbx_struct_conn_angle.ptnr1_auth_comp_id / _pdbx_struct_conn_angle.ptnr1_auth_seq_id / _pdbx_struct_conn_angle.ptnr1_label_asym_id / _pdbx_struct_conn_angle.ptnr1_label_atom_id / _pdbx_struct_conn_angle.ptnr1_label_comp_id / _pdbx_struct_conn_angle.ptnr1_label_seq_id / _pdbx_struct_conn_angle.ptnr1_symmetry / _pdbx_struct_conn_angle.ptnr3_auth_comp_id / _pdbx_struct_conn_angle.ptnr3_auth_seq_id / _pdbx_struct_conn_angle.ptnr3_label_asym_id / _pdbx_struct_conn_angle.ptnr3_label_atom_id / _pdbx_struct_conn_angle.ptnr3_label_comp_id / _pdbx_struct_conn_angle.ptnr3_label_seq_id / _pdbx_struct_conn_angle.ptnr3_symmetry / _pdbx_struct_conn_angle.value / _struct_conn.pdbx_dist_value / _struct_conn.ptnr1_auth_comp_id / _struct_conn.ptnr1_auth_seq_id / _struct_conn.ptnr1_label_asym_id / _struct_conn.ptnr1_label_atom_id / _struct_conn.ptnr1_label_comp_id / _struct_conn.ptnr1_label_seq_id / _struct_conn.ptnr1_symmetry / _struct_conn.ptnr2_auth_comp_id / _struct_conn.ptnr2_auth_seq_id / _struct_conn.ptnr2_label_asym_id / _struct_conn.ptnr2_label_atom_id / _struct_conn.ptnr2_label_comp_id / _struct_conn.ptnr2_label_seq_id / _struct_conn.ptnr2_symmetry / _struct_ref_seq_dif.details / _struct_site.pdbx_auth_asym_id / _struct_site.pdbx_auth_comp_id / _struct_site.pdbx_auth_seq_id Revision 1.5 Oct 25, 2023 Group / Refinement descriptionCategory / chem_comp_bond / pdbx_initial_refinement_model
Show all Show less
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads



















 PDBj
PDBj




 Assembly
Assembly

 Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM
Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM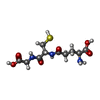
 Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S
 Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca
Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 18.015 Da / Num. of mol.: 418 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 418 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation Processing
Processing