 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1p0z | ||||||
|---|---|---|---|---|---|---|---|
| Title | Sensor Kinase CitA binding domain | ||||||
 Components Components | Sensor kinase citA | ||||||
 Keywords Keywords | TRANSFERASE / Kinase | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphorelay sensor kinase activity / histidine kinase / ATP binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  Klebsiella pneumoniae (bacteria) Klebsiella pneumoniae (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / SAD / Resolution: 1.6 Å | ||||||
 Authors Authors | Reinelt, S. / Hofmann, E. / Gerharz, T. / Bott, M. / Madden, D.R. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: The structure of the periplasmic ligand-binding domain of the sensor kinase CitA reveals the first extracellular PAS domain. Authors: Reinelt, S. / Hofmann, E. / Gerharz, T. / Bott, M. / Madden, D.R. | ||||||
| History |
| ||||||
| Remark 300 | BIOMOLECULE: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT ...BIOMOLECULE: 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 THIS ENTRY CONTAINS THE CRYSTALLOGRAPHIC ASYMMETRIC UNIT WHICH CONSISTS OF 10CHAIN(S). SEE REMARK 350 FOR INFORMATION ON GENERATING THE BIOLOGICAL MOLECULE(S). THE MONOMER IS LIGAND-BINDING COMPETENT. GEL FILTRATION ANALYSIS REVEALS WEAK DIMERIZATION IN SOLUTION WHICH MAY REPRESENT THE OLIGOMERIZATION STATE OF THE RECEPTOR IN VIVO. TWO CANDIDATE DIMER INTERFACES ARE DETECTED IN THE CRYSTAL, INVOLVING EITHER CHAINS E AND G OR CHAINS G AND J. | ||||||
| Remark 650 | HELIX determination method: Author determined | ||||||
| Remark 700 | SHEET determination method: Author determined |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1p0z.cif.gz | 317.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1p0z.ent.gz | 258.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1p0z.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/p0/1p0zftp://data.pdbj.org/pub/pdb/validation_reports/p0/1p0z | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj







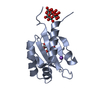
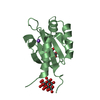
- Assembly
Assembly









-Components
-Protein , 1 types, 10 molecules ABCDEFGHIJ
| #1: Protein | Mass: 14315.185 Da / Num. of mol.: 10 / Fragment: CitA binding domain Source method: isolated from a genetically manipulated source Source: (gene. exp.) Klebsiella pneumoniae (bacteria) / Gene: CITA / Production host: |
|---|
-Non-polymers , 5 types, 1613 molecules 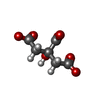

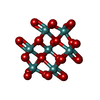


| #2: Chemical | ChemComp-FLC /  Mass: 189.100 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C6H5O7 Mass: 189.100 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: C6H5O7#3: Chemical | ChemComp-NA /  Mass: 22.990 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Na#4: Chemical | ChemComp-MO7 /  Mass: 1055.566 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Mo7O24 Mass: 1055.566 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Mo7O24#5: Chemical | ChemComp-OMO /  Mass: 145.954 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: H2MoO3 Mass: 145.954 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: H2MoO3#6: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1573 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.48 Å3/Da / Density % sol: 50.05 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 289 K / pH: 6 Details: HANGING DROP, VAPOUR DIFFUSION, RESERVOIR: 100MM NA2MOO4, 100MM MES, PH 6.0, 8% PEG4000, 2.5% GLYCEROL, PROTEIN SOLUTION: 10MM NACL, 10MM TRIS, PH 8.0, 1MM NA-CITRATE, 16MG/ML PROTEIN CONC. ...Details: HANGING DROP, VAPOUR DIFFUSION, RESERVOIR: 100MM NA2MOO4, 100MM MES, PH 6.0, 8% PEG4000, 2.5% GLYCEROL, PROTEIN SOLUTION: 10MM NACL, 10MM TRIS, PH 8.0, 1MM NA-CITRATE, 16MG/ML PROTEIN CONC. MIXING RATIO 1:1., temperature 289K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.9393 / Beamline: ID14-1 / Wavelength: 0.9393 |
| Detector | Type: ADSC QUANTUM 4r / Detector: CCD / Date: May 9, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9393 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→38.03 Å / Num. obs: 214649 / % possible obs: 95.9 % / Redundancy: 8.7 % / Biso Wilson estimate: 23.6 Å2 / Rsym value: 0.103 / Net I/σ(I): 11.88 |
| Reflection shell | Resolution: 1.6→1.7 Å / Redundancy: 8.6 % / Mean I/σ(I) obs: 4.24 / Rsym value: 0.646 / % possible all: 92.8 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Lowest resolution: 38 Å / % possible obs: 95.8 % / Redundancy: 8.7 % / Rmerge(I) obs: 0.103 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: SAD / Resolution: 1.6→38.03 Å / Rfactor Rfree error: 0.002 / Data cutoff high rms absF: 1633341.02 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: MLF Details: The high-res bin for the R_work is different from that for R_free. The bin for R_work is 1.7-1.6 A. The bin for R_free is 1.71 to 1.72. The RMS DEVIATIONS in reamrk 3 are the RMS DEVIATIONS ...Details: The high-res bin for the R_work is different from that for R_free. The bin for R_work is 1.7-1.6 A. The bin for R_free is 1.71 to 1.72. The RMS DEVIATIONS in reamrk 3 are the RMS DEVIATIONS FOR ALL ATOMS. The RMS DEVIATIONS FROM IDEAL VALUES FOR PROTEIN ATOMS are 0.014A for BOND LENGTHS, 1.70 degree for BOND ANGLES, 23.40 degree for DIHEDRAL ANGLES, 1.07 degree for IMPROPER ANGLES.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 56.47 Å2 / ksol: 0.38 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→38.03 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: RESTRAINED / Weight Biso : 2 / Weight position: 300 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.6→1.7 Å / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.6 Å / Lowest resolution: 38 Å / Rfactor Rfree: 0.19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
