 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1opl: Structural basis for the auto-inhibition of c-Abl tyrosine kinase -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1opl | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structural basis for the auto-inhibition of c-Abl tyrosine kinase | ||||||
 Components Components | proto-oncogene tyrosine-protein kinase | ||||||
 Keywords Keywords | TRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of ubiquitin-protein transferase activity / protein localization to cytoplasmic microtubule plus-end / DNA conformation change / DN4 thymocyte differentiation / response to epinephrine / phospholipase C-inhibiting G protein-coupled receptor signaling pathway / podocyte apoptotic process / transitional one stage B cell differentiation / regulation of postsynaptic specialization assembly / positive regulation of phospholipase C/protein kinase C signal transduction ...negative regulation of ubiquitin-protein transferase activity / protein localization to cytoplasmic microtubule plus-end / DNA conformation change / DN4 thymocyte differentiation / response to epinephrine / phospholipase C-inhibiting G protein-coupled receptor signaling pathway / podocyte apoptotic process / transitional one stage B cell differentiation / regulation of postsynaptic specialization assembly / positive regulation of phospholipase C/protein kinase C signal transduction / regulation of modification of synaptic structure / nicotinate-nucleotide adenylyltransferase activity / delta-catenin binding / cerebellum morphogenesis / Role of ABL in ROBO-SLIT signaling / B cell proliferation involved in immune response / neuroepithelial cell differentiation / positive regulation of Wnt signaling pathway, planar cell polarity pathway / positive regulation of extracellular matrix organization / microspike assembly / B-1 B cell homeostasis / neuropilin signaling pathway / neuropilin binding / mitochondrial depolarization / regulation of cell motility / bubble DNA binding / positive regulation of establishment of T cell polarity / cellular response to dopamine / activated T cell proliferation / positive regulation of blood vessel branching / proline-rich region binding / negative regulation of mitotic cell cycle / mitogen-activated protein kinase binding / regulation of Cdc42 protein signal transduction / regulation of hematopoietic stem cell differentiation / syntaxin binding / positive regulation of dendrite development / regulation of axon extension / regulation of T cell differentiation / alpha-beta T cell differentiation / positive regulation of cell migration involved in sprouting angiogenesis / negative regulation of cell-cell adhesion / neuromuscular process controlling balance / Myogenesis / HDR through Single Strand Annealing (SSA) / positive regulation of osteoblast proliferation / platelet-derived growth factor receptor-beta signaling pathway / RUNX2 regulates osteoblast differentiation / Fc-gamma receptor signaling pathway involved in phagocytosis / vascular endothelial cell response to oscillatory fluid shear stress / Bergmann glial cell differentiation / regulation of endocytosis / regulation of microtubule polymerization / negative regulation of long-term synaptic potentiation / negative regulation of cellular senescence / myoblast proliferation / actin monomer binding / associative learning / positive regulation of focal adhesion assembly / negative regulation of BMP signaling pathway / positive regulation of vasoconstriction / ephrin receptor signaling pathway / cardiac muscle cell proliferation / BMP signaling pathway / RHO GTPases Activate WASPs and WAVEs / cellular response to transforming growth factor beta stimulus / negative regulation of endothelial cell apoptotic process / positive regulation of T cell migration / endothelial cell migration / negative regulation of double-strand break repair via homologous recombination / regulation of cell adhesion / positive regulation of interleukin-2 production / mismatch repair / ephrin receptor binding / spleen development / ERK1 and ERK2 cascade / four-way junction DNA binding / ruffle / positive regulation of stress fiber assembly / phosphotyrosine residue binding / signal transduction in response to DNA damage / canonical NF-kappaB signal transduction / actin filament polymerization / positive regulation of substrate adhesion-dependent cell spreading / substrate adhesion-dependent cell spreading / positive regulation of mitotic cell cycle / positive regulation of endothelial cell migration / establishment of localization in cell / SH2 domain binding / Turbulent (oscillatory, disturbed) flow shear stress activates signaling by PIEZO1 and integrins in endothelial cells / response to endoplasmic reticulum stress / thymus development / protein kinase C binding / protein modification process / positive regulation of release of sequestered calcium ion into cytosol / integrin-mediated signaling pathway / regulation of autophagy / protein serine/threonine kinase activator activity / B cell receptor signaling pathway / post-embryonic development Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.42 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 3.42 Å | ||||||
 Authors Authors | Nagar, B. / Hantschel, O. / Young, M.A. / Scheffzek, K. / Veach, D. / Bornmann, W. / Clarkson, B. / Superti-Furga, G. / Kuriyan, J. | ||||||
 Citation Citation | Journal: Cell(Cambridge,Mass.) / Year: 2003 Title: Structural basis for the autoinhibition of c-Abl tyrosine kinase Authors: Nagar, B. / Hantschel, O. / Young, M.A. / Scheffzek, K. / Veach, D. / Bornmann, W. / Clarkson, B. / Superti-Furga, G. / Kuriyan, J. #1: Journal: Cell(Cambridge,Mass.) / Year: 2003Title: A myristoyl/phosphotyrosine switch regulates c-Abl Authors: Hantschel, O. / Nagar, B. / Guettler, S. / Kretzschmar, J. / Dorey, K. / Kuriyan, J. / Superti-Furga, G. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE The bound myristoyl group is from the naturally occurring N-terminal myristoyl ...SEQUENCE The bound myristoyl group is from the naturally occurring N-terminal myristoyl modification that is connected to the SH3 domain of the protein chain A by 79 residues that could not be modeled. An O atom has been intentionally omitted from MYR since the O atom is not chemically present in a myristoyl group that is attached to the protein. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1opl.cif.gz | 170.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1opl.ent.gz | 132.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1opl.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/op/1oplftp://data.pdbj.org/pub/pdb/validation_reports/op/1opl | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1opjC  1opkC  1m52S  2ablS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj









- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 61019.672 Da / Num. of mol.: 2 Fragment: N-terminal 531 residues (MYR-SH3-SH2-Kinase domain) Mutation: D382N, K29R, E29D Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: Abl / Plasmid: PFASTBAC / Production host:   Spodoptera frugiperda (fall armyworm) / References: UniProt: P00519, EC: 2.7.1.112 Spodoptera frugiperda (fall armyworm) / References: UniProt: P00519, EC: 2.7.1.112#2: Chemical | ChemComp-MYR / | 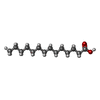  Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2 Mass: 228.371 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H28O2#3: Chemical | 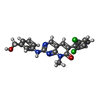  Mass: 427.283 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H16Cl2N4O2 Mass: 427.283 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H16Cl2N4O2 |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.68 Å3/Da / Density % sol: 54.13 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: 0.8 M ammonium tartrate , pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 8 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 8.2.1 / Wavelength: 1 Å / Beamline: 8.2.1 / Wavelength: 1 Å |
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jul 13, 2002 / Details: mirrors |
| Radiation | Monochromator: Double Crystal Si(111) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 3.4→75 Å / Num. all: 17250 / Num. obs: 17250 / % possible obs: 95.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 6.1 % / Biso Wilson estimate: 84.8 Å2 / Rsym value: 0.081 / Net I/σ(I): 19.7 |
| Reflection shell | Resolution: 3.4→3.52 Å / Redundancy: 5.2 % / Mean I/σ(I) obs: 3.4 / Num. unique all: 1617 / Rsym value: 0.465 / % possible all: 91.4 |
| Reflection | *PLUS Lowest resolution: 75 Å / Num. measured all: 105428 / Rmerge(I) obs: 0.081 |
| Reflection shell | *PLUS Highest resolution: 3.4 Å / % possible obs: 91.4 % / Rmerge(I) obs: 0.465 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRIES 1M52, 2ABL Resolution: 3.42→29.95 Å / Rfactor Rfree error: 0.009 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: The structure was refined by superimposing the refined high resolution structure of c-Abl (pdb entry 1OPK) on the molecular replacement solution and optimizing positions of individual ...Details: The structure was refined by superimposing the refined high resolution structure of c-Abl (pdb entry 1OPK) on the molecular replacement solution and optimizing positions of individual domains by rigid-body refinement. Following this, only overall domain B-factors were applied to molecule B, whereas individual B-factors were refined for molecule A.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 59.717 Å2 / ksol: 0.277405 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 123.3 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 3.42→29.95 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 3.4→3.61 Å / Rfactor Rfree error: 0.03 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 3.4 Å / Lowest resolution: 75 Å | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 3.4 Å / Lowest resolution: 3.52 Å |
