 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 2c0o | ||||||
|---|---|---|---|---|---|---|---|
| Title | Src family kinase Hck with bound inhibitor A-770041 | ||||||
 Components Components | TYROSINE-PROTEIN KINASE HCK | ||||||
 Keywords Keywords | TRANSFERASE / TYROSINE-PROTEIN KINASE / ATP-BINDING / KINASE / LIPOPROTEIN / MYRISTATE / NUCLEOTIDE-BINDING / PALMITATE / PHOSPHORYLATION / SH2 DOMAIN / SH3 DOMAIN | ||||||
| Function / homology |  Function and homology information Function and homology informationleukocyte degranulation / leukocyte migration involved in immune response / respiratory burst after phagocytosis / regulation of podosome assembly / innate immune response-activating signaling pathway / FLT3 signaling through SRC family kinases / regulation of phagocytosis / Nef and signal transduction / positive regulation of actin filament polymerization / Fc-gamma receptor signaling pathway involved in phagocytosis ...leukocyte degranulation / leukocyte migration involved in immune response / respiratory burst after phagocytosis / regulation of podosome assembly / innate immune response-activating signaling pathway / FLT3 signaling through SRC family kinases / regulation of phagocytosis / Nef and signal transduction / positive regulation of actin filament polymerization / Fc-gamma receptor signaling pathway involved in phagocytosis / mesoderm development / FCGR activation / type II interferon-mediated signaling pathway / transport vesicle / Signaling by CSF3 (G-CSF) / cell projection / lipopolysaccharide-mediated signaling pathway / phosphotyrosine residue binding / peptidyl-tyrosine phosphorylation / FCGR3A-mediated IL10 synthesis / integrin-mediated signaling pathway / cell surface receptor protein tyrosine kinase signaling pathway / regulation of actin cytoskeleton organization / non-specific protein-tyrosine kinase / FCGR3A-mediated phagocytosis / negative regulation of inflammatory response to antigenic stimulus / Regulation of signaling by CBL / non-membrane spanning protein tyrosine kinase activity / caveola / Inactivation of CSF3 (G-CSF) signaling / cytokine-mediated signaling pathway / cytoplasmic side of plasma membrane / Signaling by CSF1 (M-CSF) in myeloid cells / regulation of cell shape / protein autophosphorylation / regulation of inflammatory response / protein tyrosine kinase activity / cytoskeleton / protein phosphorylation / cell differentiation / lysosome / cell adhesion / intracellular signal transduction / inflammatory response / signaling receptor binding / focal adhesion / positive regulation of cell population proliferation / lipid binding / negative regulation of apoptotic process / Golgi apparatus / ATP binding / nucleus / plasma membrane / cytosol Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.85 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.85 Å | ||||||
 Authors Authors | Borhani, D.W. / Burchat, A. / Calderwood, D.J. / Hirst, G.C. / Li, B. / Loew, A. | ||||||
 Citation Citation | Journal: Bioorg.Med.Chem.Lett. / Year: 2006 Title: Discovery of A-770041, a Src-Family Selective Orally Active Lck Inhibitor that Prevents Organ Allograft Rejection. Authors: Burchat, A. / Borhani, D.W. / Calderwood, D.J. / Hirst, G.C. / Li, B. / Stachlewitz, R.F. #1: Journal: Bioorg.Med.Chem.Lett. / Year: 2004 Title: A-420983: A Potent, Orally Active Inhibitor of Lck with Efficacy in a Model of Transplant Rejection Authors: Borhani, D.W. / Calderwood, D.J. / Friedman, M.M. / Hirst, G.C. / Li, B. / Leung, A.K.W. / Mcrae, B. / Ratnofsky, S. / Ritter, K. / Waegell, W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 2c0o.cif.gz | 195.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb2c0o.ent.gz | 155.1 KB | Display | PDB format |
| PDBx/mmJSON format | 2c0o.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c0/2c0oftp://data.pdbj.org/pub/pdb/validation_reports/c0/2c0o | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  2c0iC  2c0tC  1ad5S S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |


























-Links
 PDBj
PDBj










- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Refine code: 4
NCS ensembles :
NCS oper: (Code: given Matrix: (0.939333, 0.018525, -0.342507), Vector: |
-Components
| #1: Protein | Mass: 52000.227 Da / Num. of mol.: 2 / Fragment: SH3-SH2-SH1, RESIDUES 80-525 / Mutation: YES Source method: isolated from a genetically manipulated source Details: SRC NUMBERING USED. ADD 20 TO THE RESIDUE NUMBERS IN THIS ENTRY TO OBTAIN ACTUAL HUMAN HCK RESIDUE NUMBERS. RESIDUE Y501 (HCK Y521) IS PHOSPHORYLATED Source: (gene. exp.) HOMO SAPIENS (human) / Cell: LYMPHOCYTE / Plasmid: PBC HCK002 / Cell line (production host): SF9 / Production host:   SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P08631, EC: 2.7.1.112 SPODOPTERA FRUGIPERDA (fall armyworm) / References: UniProt: P08631, EC: 2.7.1.112#2: Chemical | 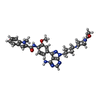  Mass: 621.732 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H39N9O3 Mass: 621.732 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H39N9O3#3: Chemical |   Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Ca#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 248 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 248 / Source method: isolated from a natural source / Formula: H2OCompound details | ENGINEERED RESIDUE IN CHAIN A, GLN 502 TO GLU ENGINEERED RESIDUE IN CHAIN A, GLN 503 TO GLU ...ENGINEERED | Has protein modification | Y | Sequence details | THE PROTEIN CRYSTALLIZED INCLUDED EIGHT NON-NATIVE N-TERMINAL RESIDUES: GLY-ALA-MET-GLY-SER-GLY-ILE- ...THE PROTEIN CRYSTALLIZ | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.1 Å3/Da / Density % sol: 59.9 % Description: INTENSITIES WERE CONVERTED TO STRUCTURE FACTORS USING CCP4 PROGRAM TRUNCATE. |
|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sitting drop / pH: 8 Details: HCK (10 MG/ML IN 150 MM NACL, 20 MM TRIS.HCL PH 8.0) WAS MIXED WITH A-770041 [100 MM STOCK SOLUTION IN DMSO) TO GIVE A FINAL A-770041 CONCENTRATION OF 1 MM. HCK/A-770041 WAS THEN MIXED WITH ...Details: HCK (10 MG/ML IN 150 MM NACL, 20 MM TRIS.HCL PH 8.0) WAS MIXED WITH A-770041 [100 MM STOCK SOLUTION IN DMSO) TO GIVE A FINAL A-770041 CONCENTRATION OF 1 MM. HCK/A-770041 WAS THEN MIXED WITH RESERVOIR SOLUTION (12% PEG 6000, 3% 1,5-DIAMINOPENTANE, 20% GLYCEROL, 200 MM CA(OAC)2, 100 MM TRIS.HCL PH 8.0) AND EQUILBRATED AGAINST THE RESERVOIR SOLUTION BY VAPOR DIFFUSION (SITTING DROPS) AT 277 K. |
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: ENRAF-NONIUS FR591 / Wavelength: 1.5418 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: May 14, 2003 / Details: OSMIC DUAL MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.85→50 Å / Num. obs: 28918 / % possible obs: 96.1 % / Observed criterion σ(I): 0 / Redundancy: 3.2 % / Biso Wilson estimate: 74.3 Å2 / Rmerge(I) obs: 0.09 / Net I/σ(I): 9.3 |
| Reflection shell | Resolution: 2.85→2.95 Å / Redundancy: 2.7 % / Rmerge(I) obs: 0.39 / Mean I/σ(I) obs: 2.5 / % possible all: 78.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1AD5 Resolution: 2.85→40 Å / Cor.coef. Fo:Fc: 0.94 / Cor.coef. Fo:Fc free: 0.892 / SU B: 32.074 / SU ML: 0.317 / TLS residual ADP flag: LIKELY RESIDUAL / Cross valid method: THROUGHOUT / ESU R Free: 0.416 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE ACTIVATION LOOP (RESIDUES A386-A398 AND B386-B398) IS DISORDERED IN BOTH HCK MOLECULES (CHAINS A AND B). HCK CHAIN A IS SLIGHTLY BETTER ...Details: HYDROGENS HAVE BEEN ADDED IN THE RIDING POSITIONS. THE ACTIVATION LOOP (RESIDUES A386-A398 AND B386-B398) IS DISORDERED IN BOTH HCK MOLECULES (CHAINS A AND B). HCK CHAIN A IS SLIGHTLY BETTER ORDERED THAN CHAIN B. SIMILARLY, THE LIGAND A1506 IS BETTER ORDERED THAN B1506. SEVEN N-TERMINAL RESIDUES (FROM THE EXPRESSION VECTOR, GLY-ALA -MET-GLY-SER-GLY-ILE) ARE DISORDERED IN BOTH CHAINS. RESIDUE ARG-59 IS NON-NATIVE (EXPRESSION VECTOR). THE HYDRATION SHELL OF CALCIUM B1507 IS POORLY DEFINED.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.2 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 51.89 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.85→40 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|