 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1obc: LEUCYL-TRNA SYNTHETASE FROM THERMUS THERMOPHILUS COMPLEXED WITH A... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1obc | ||||||
|---|---|---|---|---|---|---|---|
| Title | LEUCYL-TRNA SYNTHETASE FROM THERMUS THERMOPHILUS COMPLEXED WITH A POST-TRANSFER EDITING SUBSTRATE ANALOGUE | ||||||
 Components Components | LEUCYL-TRNA SYNTHETASE | ||||||
 Keywords Keywords | SYNTHETASE / AMINOACYL-TRNA SYNTHETASE / CLASS I AMINOACYL-TRNA SYNTHETASE / ATP + L-LEUCINE + TRNA (LEU) -> AMP + PPI L-LEUCYL-TRNA(LEU) | ||||||
| Function / homology |  Function and homology information Function and homology informationleucine-tRNA ligase / leucine-tRNA ligase activity / leucyl-tRNA aminoacylation / aminoacyl-tRNA deacylase activity / ATP binding / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |   THERMUS THERMOPHILUS (bacteria) THERMUS THERMOPHILUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.1 Å | ||||||
 Authors Authors | Cusack, S. / Yaremchuk, A. / Tukalo, M. | ||||||
 Citation Citation | Journal: Mol.Cell / Year: 2003 Title: Structural and Mechanistic Basis of Pre- and Posttransfer Editing by Leucyl-tRNA Synthetase Authors: Lincecum, T. / Tukalo, M. / Yaremchuk, A. / Mursinna, R. / Williams, A. / Sproat, B. / Van Den Eynde, W. / Link, A. / Van Calenbergh, S. / Grotli, M. / Martinis, S. / Cusack, S. #1: Journal: Embo J. / Year: 2000Title: The 2A Structure of Leucyl-tRNA Synthetase and its Complex with a Leucyl-Adenylate Analogue Authors: Cusack, S. / Yaremchuk, A. / Tukalo, M. #2: Journal: Acta Crystallogr.,Sect.D / Year: 2000 Title: Crystallization and Preliminary Crystallographic Analysis of Thermus Thermophilus Leucyl-tRNA Synthetase and its Complexes with Leucine and a Non-Hydrolysable Leucyl-Adenylate Analogue. Authors: Yaremchuk, A. / Cusack, S. / Gudzera, O. / Grotli, M. / Tukalo, M. | ||||||
| History |
| ||||||
| Remark 700 | SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN ... SHEET THE SHEET STRUCTURE OF THIS MOLECULE IS BIFURCATED. IN ORDER TO REPRESENT THIS FEATURE IN THE SHEET RECORDS BELOW, TWO SHEETS ARE DEFINED. |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1obc.cif.gz | 191.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1obc.ent.gz | 148.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1obc.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ob/1obcftp://data.pdbj.org/pub/pdb/validation_reports/ob/1obc | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1obhC  1h3nS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
-Protein , 1 types, 1 molecules A
| #1: Protein | Mass: 101170.148 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Details: ENZYME CONTAINS 878 RESIDUES AND TWO ZINC ATOMS / Source: (gene. exp.) THERMUS THERMOPHILUS (bacteria) / Strain: HB27 / Production host: |
|---|
-Non-polymers , 6 types, 493 molecules 

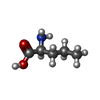


| #2: Chemical | ChemComp-LEU /  Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO2 Type: L-peptide linking / Mass: 131.173 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H13NO2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
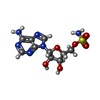
| #3: Chemical |  Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-NVA / |  Type: L-peptide linking / Mass: 117.146 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H11NO2 Type: L-peptide linking / Mass: 117.146 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H11NO2#5: Chemical | ChemComp-2AD / |  Type: L-peptide linking / Mass: 266.257 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N6O3 Type: L-peptide linking / Mass: 266.257 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N6O3#6: Chemical | ChemComp-LMS / [( |  Type: RNA linking / Mass: 346.320 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N6O6S Type: RNA linking / Mass: 346.320 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N6O6S#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 487 / Source method: isolated from a natural source / Formula: H2O |
-Details
| Sequence details | THE C-TERMINAL DOMAIN, RESIDUES 815-878, ARE DISORDERED |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.5 Å3/Da / Density % sol: 64 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: PROTEIN 10 MG/ML IN 50MM BIS-TRIS PROPANE BUFFER PH 6.0-6.5 2MM DTT, 20MM MGCL2, 1MM NAN3, 18% AMMONIUM SULPHATE EQUILBRIATED AGAINST 30-38% AMMONIUM SULPHATE IN 100MM BIS-TRIS PROPANE ...Details: PROTEIN 10 MG/ML IN 50MM BIS-TRIS PROPANE BUFFER PH 6.0-6.5 2MM DTT, 20MM MGCL2, 1MM NAN3, 18% AMMONIUM SULPHATE EQUILBRIATED AGAINST 30-38% AMMONIUM SULPHATE IN 100MM BIS-TRIS PROPANE BUFFER PH 6.0-6.5. SEE REFERENCE 2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: used microseeding, Yaremchuk, A., (2000) Acta Crystallogr., 56, 667. PH range low: 6.5 / PH range high: 6 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 / Beamline: ID14-2 / Wavelength: 0.933 |
| Detector | Type: CCD / Detector: CCD / Date: Dec 9, 2002 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.1→25 Å / Num. obs: 77419 / % possible obs: 96.2 % / Redundancy: 2.3 % / Biso Wilson estimate: 18.8 Å2 / Rmerge(I) obs: 0.088 / Net I/σ(I): 6.3 |
| Reflection shell | Resolution: 2.1→2.15 Å / Redundancy: 1.8 % / Rmerge(I) obs: 0.35 / Mean I/σ(I) obs: 2 / % possible all: 84.2 |
| Reflection shell | *PLUS % possible obs: 84.2 % / Rmerge(I) obs: 0.35 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1H3N Resolution: 2.1→24.65 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 3458288.37 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: SIDE-CHAINS ATOMS WITH NO ELECTRON DENSITY HAVE ZERO OCCUPANCY
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 49.5973 Å2 / ksol: 0.382896 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 30.8 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.1→24.65 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.1→2.23 Å / Rfactor Rfree error: 0.012 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.1 Å / Lowest resolution: 25 Å / % reflection Rfree: 4.8 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
