beta-catenin destruction complex assembly / armadillo repeat domain binding / cell development / negative regulation of glycogen (starch) synthase activity / neuron projection organization / regulation of microtubule anchoring at centrosome / negative regulation of mesenchymal stem cell differentiation / negative regulation of type B pancreatic cell development / regulation of protein export from nucleus / superior temporal gyrus development ...beta-catenin destruction complex assembly / armadillo repeat domain binding / cell development / negative regulation of glycogen (starch) synthase activity / neuron projection organization / regulation of microtubule anchoring at centrosome / negative regulation of mesenchymal stem cell differentiation / negative regulation of type B pancreatic cell development / regulation of protein export from nucleus / superior temporal gyrus development / positive regulation of protein localization to cilium / negative regulation of glycogen biosynthetic process / negative regulation of TORC2 signaling / beta-arrestin-dependent dopamine receptor signaling pathway / negative regulation of dopaminergic neuron differentiation / positive regulation of protein localization to centrosome / maintenance of cell polarity / positive regulation of cilium assembly / heart valve development / CRMPs in Sema3A signaling / tau-protein kinase / beta-catenin destruction complex / APC truncation mutants have impaired AXIN binding / AXIN missense mutants destabilize the destruction complex / Truncations of AMER1 destabilize the destruction complex / positive regulation of mitochondrial outer membrane permeabilization involved in apoptotic signaling pathway / negative regulation of calcineurin-NFAT signaling cascade / Maturation of nucleoprotein / cellular response to interleukin-3 / Beta-catenin phosphorylation cascade / Signaling by GSK3beta mutants / CTNNB1 S33 mutants aren't phosphorylated / CTNNB1 S37 mutants aren't phosphorylated / CTNNB1 S45 mutants aren't phosphorylated / CTNNB1 T41 mutants aren't phosphorylated / negative regulation of TOR signaling / I-SMAD binding / regulation of long-term synaptic potentiation / Wnt signalosome / regulation of microtubule-based process / AKT phosphorylates targets in the cytosol / Disassembly of the destruction complex and recruitment of AXIN to the membrane / regulation of axon extension / positive regulation of protein binding / regulation of neuron projection development / negative regulation of protein localization to nucleus / positive regulation of ubiquitin-dependent protein catabolic process / Maturation of nucleoprotein / glycogen metabolic process / ER overload response / RUNX1 regulates transcription of genes involved in WNT signaling / RUNX1 regulates estrogen receptor mediated transcription / negative regulation of epithelial to mesenchymal transition / positive regulation of cell-matrix adhesion / tau-protein kinase activity / regulation of axonogenesis / establishment of cell polarity / regulation of dendrite morphogenesis / Constitutive Signaling by AKT1 E17K in Cancer / protein kinase A catalytic subunit binding / SMAD binding / canonical Wnt signaling pathway / R-SMAD binding / dynactin binding / epithelial to mesenchymal transition / lateral plasma membrane / negative regulation of extrinsic apoptotic signaling pathway via death domain receptors / Regulation of HSF1-mediated heat shock response / extrinsic apoptotic signaling pathway in absence of ligand / negative regulation of osteoblast differentiation / extrinsic apoptotic signaling pathway / NF-kappaB binding / negative regulation of protein-containing complex assembly / cellular response to retinoic acid / ubiquitin-like ligase-substrate adaptor activity / regulation of cellular response to heat / positive regulation of type I interferon production / presynaptic modulation of chemical synaptic transmission / Transcriptional and post-translational regulation of MITF-M expression and activity / signaling adaptor activity / positive regulation of autophagy / positive regulation of protein export from nucleus / negative regulation of cell migration / response to endoplasmic reticulum stress / excitatory postsynaptic potential / positive regulation of protein ubiquitination / hippocampus development / protein serine/threonine kinase activator activity / cell periphery / mitochondrion organization / regulation of microtubule cytoskeleton organization / protein serine/threonine kinase binding / peptidyl-serine phosphorylation / positive regulation of cell differentiation / TCF dependent signaling in response to WNT / negative regulation of canonical Wnt signaling pathway / Ubiquitin-dependent degradation of Cyclin D / circadian rhythm / positive regulation of protein-containing complex assembly / Degradation of AXIN Similarity search - Function
Axin beta-catenin binding / Axin-1/2, tankyrase-binding domain / Axin-like / Axin beta-catenin binding motif / Axin-1 tankyrase binding domain / DIX domain / RGS, subdomain 1/3 / DIX domain superfamily / DIX domain / DIX domain profile. ...Axin beta-catenin binding / Axin-1/2, tankyrase-binding domain / Axin-like / Axin beta-catenin binding motif / Axin-1 tankyrase binding domain / DIX domain / RGS, subdomain 1/3 / DIX domain superfamily / DIX domain / DIX domain profile. / Domain present in Dishevelled and axin / Glycogen synthase kinase 3, catalytic domain / : / Regulator of G protein signaling domain / RGS domain / RGS domain profile. / Regulator of G protein signalling domain / RGS, subdomain 2 / RGS domain superfamily / Ubiquitin-like domain superfamily / Phosphorylase Kinase; domain 1 / Phosphorylase Kinase; domain 1 / Transferase(Phosphotransferase) domain 1 / Transferase(Phosphotransferase); domain 1 / Serine/threonine-protein kinase, active site / Serine/Threonine protein kinases active-site signature. / Protein kinase domain / Serine/Threonine protein kinases, catalytic domain / Protein kinase, ATP binding site / Protein kinases ATP-binding region signature. / Protein kinase domain profile. / Protein kinase domain / Protein kinase-like domain superfamily / 2-Layer Sandwich / Orthogonal Bundle / Mainly Alpha / Alpha Beta Similarity search - Domain/homology
SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW ... SHEET DETERMINATION METHOD: DSSP THE SHEETS PRESENTED AS "AA" IN EACH CHAIN ON SHEET RECORDS BELOW IS ACTUALLY AN 7-STRANDED BARREL THIS IS REPRESENTED BY A 8-STRANDED SHEET IN WHICH THE FIRST AND LAST STRANDS ARE IDENTICAL.
Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O
Has protein modification
Y
-
Experimental details
-
Experiment
Experiment
Method: X-RAY DIFFRACTION / Number of used crystals: 1
-
Sample preparation
Crystal
Density Matthews: 2.7 Å3/Da / Density % sol: 55 %
Crystal grow
Method: vapor diffusion, hanging drop / pH: 7.5 Details: CRYSTAL WERE GROWN BY THE HANGING DROP METHOD. 1UL OF PROTEIN SOLUTION (6MG/ML GSK3B AND 0.37MG/ML AXIN PEPTIDE) IN 25MM HEPES-NAOH, 250MM NACL, 1MM DTT, PH 7.0) WAS MIXED WITH 1UL ...Details: CRYSTAL WERE GROWN BY THE HANGING DROP METHOD. 1UL OF PROTEIN SOLUTION (6MG/ML GSK3B AND 0.37MG/ML AXIN PEPTIDE) IN 25MM HEPES-NAOH, 250MM NACL, 1MM DTT, PH 7.0) WAS MIXED WITH 1UL PRECIPITANT (18% PEG4000, 150MM MGCL2, 100MM TRIS- HCL, PH 7.5)
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller


 Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information HOMO SAPIENS (human)
HOMO SAPIENS (human) X-RAY DIFFRACTION /
X-RAY DIFFRACTION /  Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads
















 PDBj
PDBj







 Assembly
Assembly


 SPODOPTERA FRUGIPERDA (fall armyworm) / Strain (production host): SF9 / References: UniProt: P49841, EC: 2.7.1.37
SPODOPTERA FRUGIPERDA (fall armyworm) / Strain (production host): SF9 / References: UniProt: P49841, EC: 2.7.1.37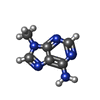
 Mass: 149.153 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H7N5
Mass: 149.153 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H7N5 Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O
Mass: 18.015 Da / Num. of mol.: 133 / Source method: isolated from a natural source / Formula: H2O Sample preparation
Sample preparation / Beamline: ID29 / Wavelength: 0.9253
/ Beamline: ID29 / Wavelength: 0.9253  Processing
Processing