 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nuv: The Leadzyme Ribozyme Bound to Mg(H2O)6(II) and Sr(II) at 1.8 A r... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nuv | ||||||
|---|---|---|---|---|---|---|---|
| Title | The Leadzyme Ribozyme Bound to Mg(H2O)6(II) and Sr(II) at 1.8 A resolution | ||||||
 Components Components |
| ||||||
 Keywords Keywords | RNA / RIBOZYME / LEADZYME / LEAD-DEPENDENT CLEAVAGE / MG(H2O)6(II) / BULGED NUCLEOTIDES / HYDRATED MAGNESIUM / SR(II) / PSEUDOHELICAL PACKING / STICKY ENDS / ALTERNATE CONFORMATION / HOMOPURINE BASE PAIRS | ||||||
| Function / homology | STRONTIUM ION / RNA / RNA (> 10) Function and homology information Function and homology information | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.81 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.81 Å | ||||||
 Authors Authors | Wedekind, J.E. / Mckay, D.B. | ||||||
 Citation Citation | Journal: BIOCHEMISTRY / Year: 2003 Title: Crystal structure of the leadzyme at 1.8 A resolution: metal ion binding and the implications for catalytic mechanism and allo site ion regulation. Authors: Wedekind, J.E. / McKay, D.B. #1: Journal: Nat.Struct.Biol. / Year: 1999Title: CRYSTAL STRUCTURE OF A LEAD DEPENDENT RIBOZYME REVEALING METAL BINDING SITES RELEVANT TO CATALYSIS Authors: Wedekind, J.E. / McKay, D.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nuv.cif.gz | 76.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nuv.ent.gz | 56.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1nuv.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nu/1nuvftp://data.pdbj.org/pub/pdb/validation_reports/nu/1nuv | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1nujC C: citing same article ( |
|---|---|
| Similar structure data | |
| Other databases |
|





-Links
 PDBj
PDBj






























- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 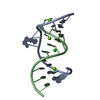
| ||||||||
| 2 | 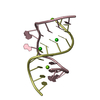
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: RNA chain | Mass: 4194.598 Da / Num. of mol.: 4 / Source method: obtained synthetically Details: THIS RIBOZYME WAS MADE BY IN VITRO SELECTION, REPORTED BY PAN & UHLENBECK #2: RNA chain | Mass: 3538.154 Da / Num. of mol.: 4 / Source method: obtained synthetically Details: THIS RIBOZYME WAS MADE BY IN VITRO SELECTION, REPORTED BY PAN & UHLENBECK #3: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Mg#4: Chemical | ChemComp-SR /   Mass: 87.620 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Sr Mass: 87.620 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Sr#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 407 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 407 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal |
| ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow |
| ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.98 Å / Beamline: BL9-1 / Wavelength: 0.98 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Mar 5, 1999 Details: 16 POLE WIGGLER, FLAT MIRROR, SI(311) BENT MONO WITH HORIZONTAL FOCUS |
| Radiation | Monochromator: SI(311) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.98 Å / Relative weight: 1 |
| Reflection | Resolution: 1.8→33.88 Å / Num. obs: 24885 / % possible obs: 94.1 % / Observed criterion σ(I): -3 / Redundancy: 2.7 % / Biso Wilson estimate: 17.4 Å2 / Rsym value: 0.035 / Net I/σ(I): 22.5 |
| Reflection shell | Resolution: 1.8→1.83 Å / Redundancy: 1.9 % / Mean I/σ(I) obs: 4.6 / Rsym value: 0.164 |
| Reflection | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 33.9 Å / Num. measured all: 228937 / Rmerge(I) obs: 0.035 |
| Reflection shell | *PLUS Rmerge(I) obs: 0.164 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: NDB ENTRY UR0001 Resolution: 1.81→28.1 Å / Rfactor Rfree error: 0.007 / Occupancy max: 1 / Occupancy min: 0.5 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||
| Solvent computation | Solvent model: CNS bulk solvent model used / Bsol: 43.3844 Å2 / ksol: 0.314622 e/Å3 | ||||||||||||||||||||
| Displacement parameters | Biso mean: 29.62 Å2
| ||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.81→28.1 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell | Resolution: 1.81→1.88 Å / Rfactor Rfree error: 0.029 / Total num. of bins used: 10
| ||||||||||||||||||||
| Software | *PLUS Name: CNS / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 33.9 Å / % reflection Rfree: 8 % / Rfactor Rfree: 0.22 / Rfactor Rwork: 0.21 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 1.86 Å / Rfactor Rfree: 0.271 / Rfactor Rwork: 0.268 |
