 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nst: THE SULFOTRANSFERASE DOMAIN OF HUMAN HAPARIN SULFATE N-DEACETYLAS... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nst | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE SULFOTRANSFERASE DOMAIN OF HUMAN HAPARIN SULFATE N-DEACETYLASE/N-SULFOTRANSFERASE | ||||||
 Components Components | HEPARAN SULFATE N-DEACETYLASE/N-SULFOTRANSFERASE | ||||||
 Keywords Keywords | SULFOTRANSFERASE / PAP / HAPARIN SULFATE / HAPARIN SULFATE BIOSYNTHESIS / GLYCOPROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology information[heparan sulfate]-glucosamine N-sulfotransferase / heparan sulfate N-sulfotransferase activity / heparan sulfate N-deacetylase activity / N-acetylglucosamine deacetylase activity / heparin proteoglycan biosynthetic process / embryonic neurocranium morphogenesis / embryonic viscerocranium morphogenesis / HS-GAG biosynthesis / glycosaminoglycan metabolic process / cardiac septum development ...[heparan sulfate]-glucosamine N-sulfotransferase / heparan sulfate N-sulfotransferase activity / heparan sulfate N-deacetylase activity / N-acetylglucosamine deacetylase activity / heparin proteoglycan biosynthetic process / embryonic neurocranium morphogenesis / embryonic viscerocranium morphogenesis / HS-GAG biosynthesis / glycosaminoglycan metabolic process / cardiac septum development / heparan sulfate proteoglycan biosynthetic process / deacetylase activity / respiratory gaseous exchange by respiratory system / coronary vasculature development / positive regulation of smoothened signaling pathway / polysaccharide biosynthetic process / aorta development / Hydrolases; Acting on carbon-nitrogen bonds, other than peptide bonds; In linear amides / midbrain development / forebrain development / fibroblast growth factor receptor signaling pathway / trans-Golgi network membrane / positive regulation of MAPK cascade / cell population proliferation / inflammatory response / Golgi membrane / Golgi apparatus Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.3 Å | ||||||
 Authors Authors | Kakuta, Y. / Pedersen, L.C. / Negishi, M. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 1999 Title: Crystal structure of the sulfotransferase domain of human heparan sulfate N-deacetylase/ N-sulfotransferase 1. Authors: Kakuta, Y. / Sueyoshi, T. / Negishi, M. / Pedersen, L.C. #1: Journal: FEBS Lett. / Year: 1998Title: A Role of Lys614 in the Sulfotransferase Activity of Human Heparan Sulfate N-Deacetylase/N-Sulfotransferase Authors: Sueyoshi, T. / Kakuta, Y. / Pedersen, L.C. / Wall, F.E. / Pedersen, L.G. / Negishi, M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nst.cif.gz | 74.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nst.ent.gz | 53.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1nst.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ns/1nstftp://data.pdbj.org/pub/pdb/validation_reports/ns/1nst | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 37917.250 Da / Num. of mol.: 1 / Fragment: SULFOTRANSFERASE DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cellular location: GOLGI MEMBRANE / Plasmid: PGEX-4T3 / Cellular location (production host): CYTOPLASM / Production host:  References: UniProt: P52848, Transferases; Transferring sulfur-containing groups; Sulfotransferases |
|---|---|
| #2: Chemical | ChemComp-A3P / 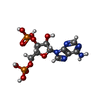  Type: RNA linking / Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 Type: RNA linking / Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 |
| #3: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 73 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 73 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.21 Å3/Da / Density % sol: 44.46 % |
|---|---|
| Crystal grow | pH: 7 / Details: pH 7.0 |
| Crystal grow | *PLUS Method: other / Details: Otwinowski, A., (1996) Methods Enzymol., 276, 307. |
-Data collection
| Diffraction | Mean temperature: 93 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH3R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Dec 4, 1997 / Details: YALE MIRRORS |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.3→50 Å / Num. obs: 13992 / % possible obs: 93.7 % / Observed criterion σ(I): 0 / Redundancy: 2.6 % / Biso Wilson estimate: 20.6 Å2 / Rsym value: 0.098 |
| Reflection shell | Resolution: 2.3→2.38 Å / Rsym value: 0.241 / % possible all: 80 |
| Reflection | *PLUS Rmerge(I) obs: 0.098 |
| Reflection shell | *PLUS % possible obs: 80 % / Rmerge(I) obs: 0.241 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2.3→50 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2 / Details: BULK SOLVENT MODEL USED
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.8 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.3→50 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.3→2.44 Å / Rfactor Rfree error: 0.031 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.21 / Rfactor Rwork: 0.21 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
