Journal: ORG.BIOMOL.CHEM. / Year: 2003 Title: Structure of the parallel-stranded DNA quadruplex d(TTAGGGT)4 containing the human telomeric repeat: evidence for A-tetrad formation from NMR and molecular dynamics simulations. Authors: Gavathiotis, E. / Searle, M.S.
Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M
Radiation wavelength
Relative weight: 1
NMR spectrometer
Type
Manufacturer
Model
Field strength (MHz)
Spectrometer-ID
Bruker DRX
Bruker
DRX
500
1
Bruker DRX
Bruker
DRX
600
2
-
Processing
NMR software
Name
Version
Classification
XwinNMR
2.6
collection
XwinNMR
2.6
processing
ANSIG
v3.3
dataanalysis
Amber
6
refinement
Refinement
Method: restrained molecular dynamics / Software ordinal: 1 Details: Structures calculated using restrained molecular dynamics.Total number of NOE restraints 728 of which 24 hydrogen-bond restraints included for the hydrogen-bonding geometry of the G-tetrads. ...Details: Structures calculated using restrained molecular dynamics.Total number of NOE restraints 728 of which 24 hydrogen-bond restraints included for the hydrogen-bonding geometry of the G-tetrads. Energy minimisations and restrained molecular dynamics were carried out using the SANDER module of AMBER 6. Calculations with SANDER were performed with a 2 fs time step, with the SHAKE algorithm (tolerance 0.00005 A) applied to all bonds to remove bond stretching, and a 9 A cut off to the Lennard Jones interactions. The restrained molecular dynamics were performed at 300K and a constant pressure of 1.0 atm with isotropic position scaling utilising the Berendsen algorithm for temperature coupling. Translational and rotational motions were removed every 100 fs. All calculations were carried out with the PME method using a 9 A cut-off for direct space non-bonded calculations and a 0.00001 Ewald convergence tolerance for the inclusion of long-range electrostatics in our calculations. The quadruplex system was allowed to equilibrate fully before the molecular dynamics calculations. Minimisation was performed with 50 steps of steepest descent and 5000 steps of conjugate gradient to first the water and counterions, with the DNA coordinates frozen, followed by a further 5000 steps on all the components of the system. Next, 10 ps unrestrained molecular dynamics were run at 100K on the water alone with the DNA and potassium ions constrained, followed for another 10 ps to allow the potassium ions to move. In the following 5 ps of dynamics the temperature of the system was increased from 100K to 300K. The next runs, each of them of 10 ps dynamics, the DNA force constant is gradually reduced from 100 to 50, 25, 10, 5 and 2.5 kcal mol-1 A-2. The equilibration step ends with 100 ps of dynamics on the whole fully unrestrained system. The system now is fully equilibrated and NOE restraints can be applied to the quadruplex system. Distance restraints were introduced gradually on the system over the first 10 ps of 100 ps MD run with the temperature stable at 300K and PME on. All NOE restraints were introduced in the form of square well potentials with a force constant of 50 kcal mol-1 A-1 for the hydrogen-bond restraints and 30 kcal mol-1 A-1 for all the other NOE distance restraints. A total of 1000 ps simulation was performed under the same conditions.Calculated structures satisfied the vast majority of the NOE restraints from the set of 728 restraints. The average minimised structure had no restraint violation > 0.3 A that contributed to a 48.22 kcal mol-1 energy penalty. Snapshots of each picosecond were extracted from the whole simulation and the structures were determined to be equilibrated on the basis of RMSD analysis.
NMR representative
Selection criteria: minimized average structure
NMR ensemble
Conformer selection criteria: structures with the lowest energy Conformers calculated total number: 100 / Conformers submitted total number: 11
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Components
Components Keywords
Keywords Function and homology information
Function and homology information Authors
Authors Citation
Citation Structure visualization
Structure visualization Downloads & links
Downloads & links Other downloads
Other downloads





 PDBj
PDBj

 Assembly
Assembly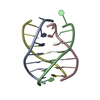
 Sample preparation
Sample preparation Processing
Processing Amber
Amber