 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1nbp: Crystal Structure Of Human Interleukin-2 Y31C Covalently Modified... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1nbp | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal Structure Of Human Interleukin-2 Y31C Covalently Modified At C31 With 3-Mercapto-1-(1,3,4,9-tetrahydro-B-carbolin-2-yl)-propan-1-one | ||||||
 Components Components | Interleukin-2 | ||||||
 Keywords Keywords | CYTOKINE / FOUR-HELIX BUNDLE / SMALL MOLECULE COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationresponse to tacrolimus / kappa-type opioid receptor binding / regulation of CD4-positive, alpha-beta T cell proliferation / regulation of T cell homeostatic proliferation / interleukin-2 receptor binding / negative regulation of lymphocyte proliferation / glycosphingolipid binding / positive regulation of plasma cell differentiation / positive regulation of tissue remodeling / negative regulation of T-helper 17 cell differentiation ...response to tacrolimus / kappa-type opioid receptor binding / regulation of CD4-positive, alpha-beta T cell proliferation / regulation of T cell homeostatic proliferation / interleukin-2 receptor binding / negative regulation of lymphocyte proliferation / glycosphingolipid binding / positive regulation of plasma cell differentiation / positive regulation of tissue remodeling / negative regulation of T-helper 17 cell differentiation / RUNX1 and FOXP3 control the development of regulatory T lymphocytes (Tregs) / leukocyte activation involved in immune response / positive regulation of isotype switching to IgG isotypes / activated T cell proliferation / Interleukin-2 signaling / cell surface receptor signaling pathway via STAT / natural killer cell activation / kinase activator activity / positive regulation of activated T cell proliferation / positive regulation of regulatory T cell differentiation / negative regulation of B cell apoptotic process / interleukin-2-mediated signaling pathway / positive regulation of immunoglobulin production / positive regulation of interleukin-17 production / positive regulation of dendritic spine development / T cell differentiation / Interleukin receptor SHC signaling / extrinsic apoptotic signaling pathway in absence of ligand / positive regulation of B cell proliferation / cytokine activity / growth factor activity / negative regulation of inflammatory response / positive regulation of type II interferon production / positive regulation of inflammatory response / cell-cell signaling / carbohydrate binding / positive regulation of cytosolic calcium ion concentration / RAF/MAP kinase cascade / positive regulation of cell growth / transcription by RNA polymerase II / phospholipase C-activating G protein-coupled receptor signaling pathway / adaptive immune response / response to ethanol / cell adhesion / immune response / positive regulation of cell population proliferation / negative regulation of apoptotic process / positive regulation of transcription by RNA polymerase II / : / extracellular region Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Hyde, J. / Braisted, A.C. / Randal, M. / Arkin, M.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2003 Title: Discovery and characterization of cooperative ligand binding in the adaptive region of interleukin-2 Authors: Hyde, J. / Braisted, A.C. / Randal, M. / Arkin, M.R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1nbp.cif.gz | 39.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1nbp.ent.gz | 26.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1nbp.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/nb/1nbpftp://data.pdbj.org/pub/pdb/validation_reports/nb/1nbp | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1m4aS S: Starting model for refinement |
|---|---|
| Similar structure data |















-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 15375.948 Da / Num. of mol.: 1 / Mutation: Y31C Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: IL2 / Plasmid: pRSET / Species (production host): Escherichia coli / Production host:  |
|---|---|
| #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 |
| #3: Chemical | ChemComp-MHC / 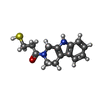  Mass: 260.355 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H16N2OS Mass: 260.355 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C14H16N2OS |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 49 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.34 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 7.5 Details: ammonium sulfate, PEG 8k, pH 7.5, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||
| Crystal grow | *PLUS | |||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Date: Aug 17, 2001 / Details: mirrors |
| Radiation | Monochromator: yale miorrors / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→20 Å / Num. obs: 7096 / % possible obs: 98.2 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 3.1 % / Biso Wilson estimate: 33 Å2 / Rmerge(I) obs: 0.069 / Rsym value: 0.069 / Net I/σ(I): 17.5 |
| Reflection shell | Resolution: 2.2→2.28 Å / Redundancy: 3.1 % / Rmerge(I) obs: 0.253 / Mean I/σ(I) obs: 5.5 / Num. unique all: 715 / Rsym value: 0.253 / % possible all: 96.9 |
| Reflection | *PLUS Lowest resolution: 10 Å |
| Reflection shell | *PLUS % possible obs: 96.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1M4A Resolution: 2.2→10 Å / Cor.coef. Fo:Fc: 0.928 / Cor.coef. Fo:Fc free: 0.893 / SU B: 6.077 / SU ML: 0.177 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / ESU R: 0.338 / ESU R Free: 0.249 / Stereochemistry target values: MAXIMUM LIKELIHOOD
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 28.507 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→10 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.273 Å / Total num. of bins used: 15 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 10 Å / Rfactor Rfree: 0.277 / Rfactor Rwork: 0.228 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
