 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n7b | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Title | RIP-Radiation-damage Induced Phasing | ||||||||||||||||||
 Components Components | RNA/DNA (5'-R(* Keywords KeywordsDNA / RNA | Function / homology | : / SPERMINE / DNA/RNA hybrid |  Function and homology information Function and homology informationMethod |  X-RAY DIFFRACTION / SYNCHROTRON / RIP - Radiation-damage Induced Phasing / Resolution: 1.4 Å X-RAY DIFFRACTION / SYNCHROTRON / RIP - Radiation-damage Induced Phasing / Resolution: 1.4 Å  Authors AuthorsRavelli, R.B.G. / Leiros, H.-K.S. / Pan, B. / Caffrey, M. / McSweeney, S. |  CitationJournal: Structure / Year: 2003 CitationJournal: Structure / Year: 2003Title: Specific Radiation-Damage Can Be Used To Solve Macromolecular Crystal Structures Authors: Ravelli, R.B.G. / Leiros, H.-K.S. / Pan, B. / Caffrey, M. / McSweeney, S. History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n7b.cif.gz | 70 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n7b.ent.gz | 54.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1n7b.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n7/1n7bftp://data.pdbj.org/pub/pdb/validation_reports/n7/1n7b | HTTPS FTP |
|---|
-Related structure data








-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: DNA/RNA hybrid | Mass: 1995.093 Da / Num. of mol.: 8 / Source method: obtained synthetically Details: The RNA hexamer UGAGGU where the 2nd G was replaced by a C8-brominated Gd was phased using radiation damage on the highly susceptible Br-C8 bond. #2: Chemical | ChemComp-K /   Mass: 39.098 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 9 / Source method: obtained synthetically / Formula: K#3: Chemical | 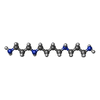  Mass: 202.340 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H26N4 Mass: 202.340 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H26N4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 181 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 181 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.77 Å3/Da / Density % sol: 30.64 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop / pH: 6 Details: 1 mM (single strand) RNA, 20 mM sodium cacodylate buffer, pH 6.0, 5 mM spermine tetrachloride, 32 mM KCl and 2 % (v/v) methyl-2,4-pentanediol (MPD) against 25 % MPD in the reservoir, VAPOR ...Details: 1 mM (single strand) RNA, 20 mM sodium cacodylate buffer, pH 6.0, 5 mM spermine tetrachloride, 32 mM KCl and 2 % (v/v) methyl-2,4-pentanediol (MPD) against 25 % MPD in the reservoir, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions |
| ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / pH: 6 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-2 / Wavelength: 0.933 Å / Beamline: ID14-2 / Wavelength: 0.933 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Feb 8, 2002 |
| Radiation | Monochromator: diamond(111) and Ge(200) / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 1.4→20 Å / Num. all: 22939 / Num. obs: 22939 / % possible obs: 99.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): -3 / Redundancy: 4.25 % / Biso Wilson estimate: 6.4 Å2 / Rmerge(I) obs: 0.095 / Net I/σ(I): 10.99 |
| Reflection shell | Resolution: 1.4→1.48 Å / Redundancy: 4.24 % / Rmerge(I) obs: 0.465 / Mean I/σ(I) obs: 3.1 / Num. unique all: 3438 / % possible all: 98.1 |
| Reflection | *PLUS Highest resolution: 1.4 Å / Lowest resolution: 20 Å / Redundancy: 4.2 % |
| Reflection shell | *PLUS Highest resolution: 1.4 Å / % possible obs: 98.1 % / Redundancy: 4.2 % / Rmerge(I) obs: 0.464 / Mean I/σ(I) obs: 3.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: RIP - Radiation-damage Induced Phasing Resolution: 1.4→20 Å / Cor.coef. Fo:Fc: 0.96 / Cor.coef. Fo:Fc free: 0.936 / SU B: 0.986 / SU ML: 0.039 Isotropic thermal model: The DNA/RNA and K-atoms were refined anisotropically, the spermine and waters isotropic Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): -3 / ESU R: 0.071 / ESU R Free: 0.068 / Stereochemistry target values: MAXIMUM LIKELIHOOD Details: The model was built and refined against the data set collected before the "X-ray burn". Afterwards, the model was refined for some additional cycles against the data set that was collected ...Details: The model was built and refined against the data set collected before the "X-ray burn". Afterwards, the model was refined for some additional cycles against the data set that was collected after the "X-ray burn". No manual adjustments were made, resulting in strong negative densities in the Fo-Fc difference map on X-ray susceptible sites, such as the Br-atoms.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 9.645 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.4→1.476 Å / Total num. of bins used: 10 /
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.42 Å / Lowest resolution: 20 Å / Rfactor Rfree: 0.199 / Rfactor Rwork: 0.152 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.4 Å / Lowest resolution: 1.48 Å |
