 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1n1i | ||||||
|---|---|---|---|---|---|---|---|
| Title | The structure of MSP-1(19) from Plasmodium knowlesi | ||||||
 Components Components | Merozoite surface protein-1 | ||||||
 Keywords Keywords | CELL ADHESION / MSP1 / malaria / surface protein / surface antigen / glycoprotein / EGF domain | ||||||
| Function / homology |  Function and homology information Function and homology information | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.4 Å | ||||||
 Authors Authors | Garman, S.C. / Simcoke, W.N. / Stowers, A.W. / Garboczi, D.N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: Structure of the C-terminal domains of merozoite surface protein-1 from Plasmodium knowlesi reveals a novel histidine binding site Authors: Garman, S.C. / Simcoke, W.N. / Stowers, A.W. / Garboczi, D.N. | ||||||
| History |
| ||||||
| Remark 999 | SEQUENCE The first five residues of the crystallized protein (GLU-ALA-GLU-ALA-SER) are non-native; ...SEQUENCE The first five residues of the crystallized protein (GLU-ALA-GLU-ALA-SER) are non-native; they are the remains of the yeast alpha mating factor secretory signal |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1n1i.cif.gz | 89 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1n1i.ent.gz | 67.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1n1i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1n1i_validation.pdf.gz | 477.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1n1i_full_validation.pdf.gz | 482.9 KB | Display | |
| Data in XML | 1n1i_validation.xml.gz | 22.5 KB | Display | |
| Data in CIF | 1n1i_validation.cif.gz | 30.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/n1/1n1iftp://data.pdbj.org/pub/pdb/validation_reports/n1/1n1i | HTTPS FTP |
-Related structure data
| Related structure data |  1b9wS S: Starting model for refinement |
|---|---|
| Similar structure data |







-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | There are four copies of the biological monomer in the asymmetric unit. |
-Components
| #1: Protein | Mass: 11450.643 Da / Num. of mol.: 4 / Fragment: C-terminal EGF-LIKE DOMAINS Source method: isolated from a genetically manipulated source Source: (gene. exp.) Species: Plasmodium knowlesi / Strain: MALAYAN H / Gene: MSP1 / Plasmid: YEpRPEU-3 / Production host:  #2: Chemical |   Mass: 69.085 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H5N2#3: Chemical | ChemComp-HIS / | 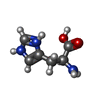  Type: L-peptide linking / Mass: 156.162 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10N3O2 Type: L-peptide linking / Mass: 156.162 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C6H10N3O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 306 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 306 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 68 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 7 Details: PEG 6000, HEPES, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU / Detector: IMAGE PLATE / Date: Feb 23, 2000 / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.4→50 Å / Num. all: 16555 / Num. obs: 16555 / % possible obs: 97.3 % / Observed criterion σ(F): 0 / Redundancy: 3.4 % / Biso Wilson estimate: 47.8 Å2 / Rmerge(I) obs: 0.082 / Rsym value: 0.082 / Net I/σ(I): 14.9 |
| Reflection shell | Resolution: 2.4→2.49 Å / Redundancy: 3 % / Rmerge(I) obs: 0.315 / Mean I/σ(I) obs: 3.4 / Num. unique all: 1565 / Rsym value: 0.315 / % possible all: 93.7 |
| Reflection | *PLUS Lowest resolution: 50 Å / Num. obs: 16570 / % possible obs: 97.7 % / Num. measured all: 56495 |
| Reflection shell | *PLUS Highest resolution: 2.4 Å / % possible obs: 93.7 % / Num. unique obs: 1565 / Num. measured obs: 4653 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1B9W Resolution: 2.4→20.59 Å / Rfactor Rfree error: 0.009 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: ENGH & HUBER Details: 300 KCAL/MOL/A^2 NCS RESTRAINTS APPLIED TO ALL ATOMS IN EARLY ROUNDS OF REFINEMENT AND RELAXED IN LATER ROUNDS.
| ||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 40.5311 Å2 / ksol: 0.257708 e/Å3 | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 55.4 Å2
| ||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.4→20.59 Å
| ||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.4→2.55 Å / Rfactor Rfree error: 0.036 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.4 Å / Lowest resolution: 50 Å | ||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
