 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bte: CRYSTAL STRUCTURE OF THE EXTRACELLULAR DOMAIN OF THE TYPE II ACTI... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bte | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE EXTRACELLULAR DOMAIN OF THE TYPE II ACTIVIN RECEPTOR | |||||||||
 Components Components | PROTEIN (ACTIVIN RECEPTOR TYPE II) | |||||||||
 Keywords Keywords | TRANSFERASE / RECEPTOR / SERINE KINASE / LIGAND BINDING DOMAIN / THREE-FINGER TOXIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationTGFBR3 regulates activin signaling / Signaling by Activin / penile erection / inhibin binding / inhibin-betaglycan-ActRII complex / activin receptor activity / Signaling by BMP / activin receptor activity, type II / positive regulation of activin receptor signaling pathway / positive regulation of follicle-stimulating hormone secretion ...TGFBR3 regulates activin signaling / Signaling by Activin / penile erection / inhibin binding / inhibin-betaglycan-ActRII complex / activin receptor activity / Signaling by BMP / activin receptor activity, type II / positive regulation of activin receptor signaling pathway / positive regulation of follicle-stimulating hormone secretion / Sertoli cell proliferation / cellular response to oxygen-glucose deprivation / sperm ejaculation / BMP receptor activity / embryonic skeletal system development / activin receptor activity, type I / activin receptor complex / receptor protein serine/threonine kinase / activin binding / cellular response to BMP stimulus / pattern specification process / SMAD protein signal transduction / activin receptor signaling pathway / gastrulation with mouth forming second / regulation of nitric oxide biosynthetic process / determination of left/right symmetry / negative regulation of ossification / anterior/posterior pattern specification / growth factor binding / odontogenesis of dentin-containing tooth / mesoderm development / positive regulation of SMAD protein signal transduction / regulation of signal transduction / positive regulation of osteoblast differentiation / positive regulation of bone mineralization / BMP signaling pathway / coreceptor activity / positive regulation of erythrocyte differentiation / PDZ domain binding / cellular response to growth factor stimulus / male gonad development / autophagy / osteoblast differentiation / spermatogenesis / signaling receptor complex / protein serine/threonine kinase activity / cell surface / positive regulation of transcription by RNA polymerase II / ATP binding / metal ion binding / identical protein binding / plasma membrane / cytoplasm Similarity search - Function | |||||||||
| Biological species |  | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MIRAS / Resolution: 1.5 Å X-RAY DIFFRACTION / SYNCHROTRON / MIRAS / Resolution: 1.5 Å | |||||||||
 Authors Authors | Greenwald, J. / Fischer, W. / Vale, W. / Choe, S. | |||||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1999 Title: Three-finger toxin fold for the extracellular ligand-binding domain of the type II activin receptor serine kinase. Authors: Greenwald, J. / Fischer, W.H. / Vale, W.W. / Choe, S. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bte.cif.gz | 57 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bte.ent.gz | 40.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1bte.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bt/1bteftp://data.pdbj.org/pub/pdb/validation_reports/bt/1bte | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 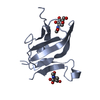
| ||||||||||||
| 2 | 
| ||||||||||||
| Unit cell |
| ||||||||||||
| Components on special symmetry positions |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.8945, -0.4458, -0.0349), Vector: |
-Components
| #1: Protein | Mass: 11507.803 Da / Num. of mol.: 2 / Fragment: LIGAND-BINDING DOMAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.)  Pichia pastoris (fungus) / Strain (production host): SMD1168 Pichia pastoris (fungus) / Strain (production host): SMD1168References: UniProt: P27038, Transferases; Transferring phosphorus-containing groups; Phosphotransferases with an alcohol group as acceptor #2: Sugar | ChemComp-NAG /   Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4 Type: D-saccharide, beta linking / Mass: 221.208 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C8H15NO6 #3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 202 / Source method: isolated from a natural source / Formula: H2OCompound details | GLYCOSYLATION FROM HOST EXPRESSION SYSTEM WAS REMOVED USING ENDOGLYCOSIDASE H, LEAVING BEHIND THE N- ...GLYCOSYLAT | Has protein modification | Y | Nonpolymer details | O6 OF NAG A 124 AND NAG B 124 IS MODELED IN TWO ALTERNATIV | Sequence details | THE NUMBERING OF RESIDUES IN THE PDB FILE IS BASED ON THE SEQUENCE WITHOUT THE SIGNAL PEPTIDE. THE ...THE NUMBERING OF RESIDUES IN THE PDB FILE IS BASED ON THE SEQUENCE WITHOUT THE SIGNAL PEPTIDE. THE MOLECULE IN THE CRYSTAL IS LACKING THE 14 C-TERMINAL RESIDUES WHICH HAVE BEEN REMOVED BY TREATMENT WITH ENDOPROTEI | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.4 Å3/Da / Density % sol: 42 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 4.5 Details: 100 MM SODIUM ACETATE, PH 4.5, 5% PEG 8000, 0.5M NACL | ||||||||||||||||||||||||
| Crystal | *PLUS Density % sol: 43 % | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 0.97 / Beamline: 5.0.2 / Wavelength: 0.97 |
| Detector | Type: ADSC / Detector: CCD / Date: Jan 15, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97 Å / Relative weight: 1 |
| Reflection | Resolution: 1.5→50 Å / Num. obs: 30005 / % possible obs: 83.9 % / Redundancy: 4.2 % / Biso Wilson estimate: 18.1 Å2 / Rsym value: 0.034 / Net I/σ(I): 11.6 |
| Reflection shell | Resolution: 1.5→1.54 Å / Redundancy: 2.9 % / Mean I/σ(I) obs: 0.3 / Rsym value: 0.295 / % possible all: 37.3 |
| Reflection | *PLUS Rmerge(I) obs: 0.034 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MIRAS / Resolution: 1.5→20 Å / SU ML: 0.063 / σ(F): 0 / ESU R: 0.086 / ESU R Free: 0.092
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.5→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.5 Å / σ(F): 0 / % reflection Rfree: 5 % / Rfactor obs: 0.181 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 23.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
