 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1md3: A folding mutant of human class pi glutathione transferase, creat... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1md3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | A folding mutant of human class pi glutathione transferase, created by mutating glycine 146 of the wild-type protein to alanine | ||||||
 Components Components | pi glutathione transferase | ||||||
 Keywords Keywords | TRANSFERASE / GST / nucleation mechanism / conserved folding module | ||||||
| Function / homology |  Function and homology information Function and homology informationnitric oxide storage / S-nitrosoglutathione binding / cellular response to cell-matrix adhesion / TRAF2-GSTP1 complex / negative regulation of smooth muscle cell chemotaxis / negative regulation of leukocyte proliferation / dinitrosyl-iron complex binding / common myeloid progenitor cell proliferation / hepoxilin biosynthetic process / glutathione derivative biosynthetic process ...nitric oxide storage / S-nitrosoglutathione binding / cellular response to cell-matrix adhesion / TRAF2-GSTP1 complex / negative regulation of smooth muscle cell chemotaxis / negative regulation of leukocyte proliferation / dinitrosyl-iron complex binding / common myeloid progenitor cell proliferation / hepoxilin biosynthetic process / glutathione derivative biosynthetic process / response to L-ascorbic acid / linoleic acid metabolic process / Glutathione conjugation / negative regulation of monocyte chemotactic protein-1 production / nitric oxide binding / JUN kinase binding / glutathione peroxidase activity / oligodendrocyte development / Paracetamol ADME / negative regulation of stress-activated MAPK cascade / negative regulation of JNK cascade / prostaglandin metabolic process / cellular response to glucocorticoid stimulus / negative regulation of interleukin-1 beta production / regulation of stress-activated MAPK cascade / Detoxification of Reactive Oxygen Species / negative regulation of acute inflammatory response / glutathione transferase / animal organ regeneration / negative regulation of ferroptosis / response to amino acid / negative regulation of vascular associated smooth muscle cell proliferation / glutathione transferase activity / negative regulation of tumor necrosis factor production / negative regulation of fibroblast proliferation / protein serine/threonine kinase inhibitor activity / regulation of ERK1 and ERK2 cascade / negative regulation of tumor necrosis factor-mediated signaling pathway / toxic substance binding / negative regulation of MAPK cascade / positive regulation of superoxide anion generation / xenobiotic metabolic process / cellular response to epidermal growth factor stimulus / negative regulation of extrinsic apoptotic signaling pathway / fatty acid binding / negative regulation of canonical NF-kappaB signal transduction / glutathione metabolic process / response to reactive oxygen species / central nervous system development / negative regulation of ERK1 and ERK2 cascade / cellular response to insulin stimulus / response to estradiol / cellular response to lipopolysaccharide / secretory granule lumen / vesicle / ficolin-1-rich granule lumen / response to ethanol / Neutrophil degranulation / negative regulation of apoptotic process / negative regulation of transcription by RNA polymerase II / mitochondrion / : / extracellular exosome / extracellular region / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.03 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.03 Å | ||||||
 Authors Authors | Kong, G.K.-W. / Dragani, B. / Aceto, A. / Cocco, R. / Mannervik, B. / Stenberg, G. / McKinstry, W.J. / Polekhina, G. / Parker, M.W. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2003 Title: Contribution of Glycine 146 to a Conserved Folding Module Affecting Stability and Refolding of Human Glutathione Transferase P1-1 Authors: Kong, G.K.-W. / Polekhina, G. / McKinstry, W.J. / Parker, M.W. / Dragani, B. / Aceto, A. / Paludi, D. / Principe, D.R. / Mannervik, B. / Stenberg, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1md3.cif.gz | 104.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1md3.ent.gz | 79.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1md3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/md/1md3ftp://data.pdbj.org/pub/pdb/validation_reports/md/1md3 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1md4C  10gsS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |



















-Links
 PDBj
PDBj




- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Components on special symmetry positions |
|
-Components
| #1: Protein | Mass: 23260.598 Da / Num. of mol.: 2 / Mutation: G145A Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: pKHP1 / Production host:  #2: Chemical |   Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM Mass: 195.237 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13NO4S / Comment: pH buffer*YM#3: Chemical | 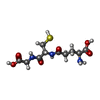  Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S Mass: 307.323 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C10H17N3O6S#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 423 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 423 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.58 Å3/Da / Density % sol: 52.33 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: vapor diffusion, hanging drop / pH: 5.6 Details: MES, PEG 8000, calcium chloride, DTT(dithiothreitol), glutathione(reduced), pH 5.6, VAPOR DIFFUSION, HANGING DROP, temperature 295K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 22 ℃ / pH: 7 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.54179 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Aug 1, 2001 / Details: mirror |
| Radiation | Monochromator: nickel / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54179 Å / Relative weight: 1 |
| Reflection | Resolution: 2.03→20 Å / Num. all: 30540 / Num. obs: 29161 / % possible obs: 95.5 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 2.7 % / Biso Wilson estimate: 16.5 Å2 / Rmerge(I) obs: 0.057 |
| Reflection shell | Resolution: 2.03→2.1 Å / Rmerge(I) obs: 0.273 / % possible all: 85.8 |
| Reflection | *PLUS Highest resolution: 2.03 Å / Num. obs: 29161 / % possible obs: 95.5 % / Num. measured all: 77558 / Rmerge(I) obs: 0.057 |
| Reflection shell | *PLUS % possible obs: 85.8 % / Mean I/σ(I) obs: 3 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 10GS Resolution: 2.03→19.38 Å / Rfactor Rfree error: 0.006 / Data cutoff high absF: 1569797.24 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 53.5238 Å2 / ksol: 0.35151 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 23.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.03→19.38 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS | NCS model details: CONSTR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.03→2.1 Å / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.03 Å / Lowest resolution: 19.38 Å / Num. reflection obs: 27705 / Num. reflection Rfree: 1454 / % reflection Rfree: 5 % / Rfactor Rfree: 0.218 / Rfactor Rwork: 0.181 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
