 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1lgn | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | DECAMERIC DAMP COMPLEX OF HUMAN SERUM AMYLOID P COMPONENT | |||||||||
 Components Components | SERUM AMYLOID P COMPONENT | |||||||||
 Keywords Keywords | SERUM PROTEIN / AMYLOIDOSIS / DRUG DESIGN / NUCLEOTIDE | |||||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation by host of viral glycoprotein metabolic process / negative regulation of glycoprotein metabolic process / complement component C1q complex binding / negative regulation of viral process / negative regulation of wound healing / negative regulation of monocyte differentiation / host-mediated suppression of symbiont invasion / virion binding / negative regulation of acute inflammatory response / chaperone-mediated protein complex assembly ...negative regulation by host of viral glycoprotein metabolic process / negative regulation of glycoprotein metabolic process / complement component C1q complex binding / negative regulation of viral process / negative regulation of wound healing / negative regulation of monocyte differentiation / host-mediated suppression of symbiont invasion / virion binding / negative regulation of acute inflammatory response / chaperone-mediated protein complex assembly / acute-phase response / : / carbohydrate binding / protein folding / blood microparticle / Amyloid fiber formation / innate immune response / calcium ion binding / : / extracellular exosome / extracellular region / identical protein binding / nucleus Similarity search - Function | |||||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | |||||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | |||||||||
 Authors Authors | Hohenester, E. / Pepys, M.B. / Wood, S.P. | |||||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 1997 Title: Crystal structure of a decameric complex of human serum amyloid P component with bound dAMP. Authors: Hohenester, E. / Hutchinson, W.L. / Pepys, M.B. / Wood, S.P. #1: Journal: Nature / Year: 1994Title: Structure of Pentameric Human Serum Amyloid P Component Authors: Emsley, J. / White, H.E. / O'Hara, B.P. / Oliva, G. / Srinivasan, N. / Tickle, I.J. / Blundell, T.L. / Pepys, M.B. / Wood, S.P. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1lgn.cif.gz | 216.2 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1lgn.ent.gz | 173.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1lgn.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/lg/1lgnftp://data.pdbj.org/pub/pdb/validation_reports/lg/1lgn | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1sacS S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| |||||||||||||||||||||||||||||||||||
| Unit cell |
| |||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS oper:
| |||||||||||||||||||||||||||||||||||
| Details | SAP IS A PENTAMER OF IDENTICAL SUBUNITS. |
-Components
| #1: Protein | Mass: 23282.455 Da / Num. of mol.: 5 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / Tissue: SERUM / References: UniProt: P02743#2: Chemical | ChemComp-CA /   Mass: 40.078 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 10 / Source method: obtained synthetically / Formula: Ca#3: Chemical | ChemComp-D5M / 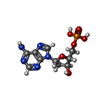  Mass: 331.222 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H14N5O6P Mass: 331.222 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: C10H14N5O6PHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 4.24 Å3/Da / Density % sol: 71 % Description: THE DIFFRACTION LIMIT IS VERY ANISOTROPIC (BETTER THAN 2.8 A ALONG A* AND B*, APPROXIMATELY 3.5 A ALONG C*) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8 Details: 3 + 3 UL HANGING DROPS AT 277K PROTEIN SOLUTION: 20 MG/ML PROTEIN, 10 MM TRIS-HCL, PH 8.0, 140 MM NACL, 20 MM DAMP, 0.02 % NA-AZIDE. RESERVOIR SOLUTION: 100 MM BIS-TRIS PROPANE- HCL PH 8.0, ...Details: 3 + 3 UL HANGING DROPS AT 277K PROTEIN SOLUTION: 20 MG/ML PROTEIN, 10 MM TRIS-HCL, PH 8.0, 140 MM NACL, 20 MM DAMP, 0.02 % NA-AZIDE. RESERVOIR SOLUTION: 100 MM BIS-TRIS PROPANE- HCL PH 8.0, 20 MM CALCIUM CHLORIDE, 12-14 % (W/V) POLYETHYLENEGLYCOL 4000., vapor diffusion - hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion, hanging dropDetails: drop solution was mixed with an equal volume of reservoir solution | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 277 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jul 1, 1996 |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2.8→30 Å / Num. obs: 366263 / % possible obs: 98 % / Redundancy: 6.9 % / Biso Wilson estimate: 62 Å2 / Rmerge(I) obs: 0.085 / Net I/σ(I): 6.8 |
| Reflection shell | Resolution: 2.8→2.9 Å / Redundancy: 6.9 % / Rmerge(I) obs: 0.443 / Mean I/σ(I) obs: 1.4 / % possible all: 97.5 |
| Reflection | *PLUS Num. obs: 53133 / Num. measured all: 366263 / Rmerge(I) obs: 0.068 |
| Reflection shell | *PLUS % possible obs: 97.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1SAC WITHOUT WATER MOLECULES AND ACETATE IONS, ALL B-FACTORS SET TO 40 A**2 Resolution: 2.8→8 Å / Data cutoff high absF: 1000000 / Data cutoff low absF: 0 / Isotropic thermal model: SEE REMARKS / Cross valid method: FREE R / σ(F): 0 Details: 15 THIN RESOLUTION SHELLS WERE DEFINED FOR THE CALCULATION OF THE FREE R VALUE USING XDLDATAMAN (CCP4).
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 50 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati sigma a obs: 0.51 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints NCS |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.92 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.454 |
