 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kt8: HUMAN BRANCHED CHAIN AMINO ACID AMINOTRANSFERASE (MITOCHONDRIAL):... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kt8 | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN BRANCHED CHAIN AMINO ACID AMINOTRANSFERASE (MITOCHONDRIAL): THREE DIMENSIONAL STRUCTURE OF ENZYME IN ITS KETIMINE FORM WITH THE SUBSTRATE L-ISOLEUCINE | ||||||
 Components Components | BRANCHED-CHAIN AMINO ACID AMINOTRANSFERASE, MITOCHONDRIAL | ||||||
 Keywords Keywords | TRANSFERASE / FOLD TYPE IV | ||||||
| Function / homology |  Function and homology information Function and homology informationregulation of hormone levels / branched-chain-amino-acid:2-oxoglutarate transaminase activity / L-valine:2-oxoglutarate transaminase activity / L-isoleucine:2-oxoglutarate transaminase activity / L-leucine:2-oxoglutarate transaminase activity / L-isoleucine catabolic process / branched-chain amino acid biosynthetic process / branched-chain-amino-acid transaminase / L-leucine biosynthetic process / Branched-chain amino acid catabolism ...regulation of hormone levels / branched-chain-amino-acid:2-oxoglutarate transaminase activity / L-valine:2-oxoglutarate transaminase activity / L-isoleucine:2-oxoglutarate transaminase activity / L-leucine:2-oxoglutarate transaminase activity / L-isoleucine catabolic process / branched-chain amino acid biosynthetic process / branched-chain-amino-acid transaminase / L-leucine biosynthetic process / Branched-chain amino acid catabolism / branched-chain amino acid catabolic process / L-valine biosynthetic process / brown fat cell differentiation / cellular response to leukemia inhibitory factor / lipid metabolic process / mitochondrial matrix / mitochondrion / nucleoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Yennawar, N.H. / Conway, M.E. / Yennawar, H.P. / Farber, G.K. / Hutson, S.M. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2002 Title: Crystal structures of human mitochondrial branched chain aminotransferase reaction intermediates: ketimine and pyridoxamine phosphate forms Authors: Yennawar, N.H. / Conway, M.E. / Yennawar, H.P. / Farber, G.K. / Hutson, S.M. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2001Title: The Structure of Human Mitochondrial Branched-Chain Aminotransferase Authors: Yennawar, N.H. / Dunbar, J. / Conway, M. / Hutson, S.M. / Farber, G.K. #2: Journal: Biochim.Biophys.Acta / Year: 1997Title: Cloning of the Rat and Human Mitochondrial Branched Amino Acid Aminotransferase (Bcatm). Authors: Bledsoe, R.K. / Dawson, P.A. / Hutson, S.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kt8.cif.gz | 162.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kt8.ent.gz | 128.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1kt8.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kt/1kt8ftp://data.pdbj.org/pub/pdb/validation_reports/kt/1kt8 | HTTPS FTP |
|---|
-Related structure data




















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41368.016 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Plasmid: PET-28A / Species (production host): Escherichia coli / Production host:  References: UniProt: O15382, branched-chain-amino-acid transaminase #2: Chemical | 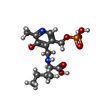  Mass: 362.315 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H23N2O7P Mass: 362.315 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C14H23N2O7P#3: Chemical | ChemComp-ACY /   Mass: 60.052 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H4O2 Mass: 60.052 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: C2H4O2#4: Chemical | ChemComp-GOL / |   Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3 Mass: 92.094 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C3H8O3#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 221 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 221 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.38 Å3/Da / Density % sol: 48.25 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 / Details: PEG1500, HEPES, DTT, pH 7.0 | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: Yennawar, N., (2001) Acta Crystallogr., Sect.D, 57, 506. | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 223 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 1.0721203 / Beamline: 5.0.2 / Wavelength: 1.0721203 |
| Detector | Detector: CCD / Date: Apr 20, 1999 |
| Radiation | Monochromator: SI(111) DOUBLE CRYSTAL / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.0721203 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→19.72 Å / Num. all: 61893 / Num. obs: 60100 / % possible obs: 95.6 % / Observed criterion σ(I): 0 / Redundancy: 4.7 % / Biso Wilson estimate: 21.3 Å2 / Rmerge(I) obs: 0.069 / Net I/σ(I): 5.4 |
| Reflection shell | Resolution: 1.9→1.93 Å / Redundancy: 4.7 % / Rmerge(I) obs: 0.66 / % possible all: 41 |
| Reflection | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 19.7 Å / Num. obs: 61939 / Redundancy: 4.7 % / Num. measured all: 238575 |
| Reflection shell | *PLUS % possible obs: 90.3 % / Rmerge(I) obs: 0.666 / Mean I/σ(I) obs: 0.9 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 1.9→19.72 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 2356223.4 / Data cutoff high rms absF: 2356223.4 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 40.197 Å2 / ksol: 0.320221 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 37.2 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→19.72 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 1.9→2.02 Å / Rfactor Rfree error: 0.015 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 19.7 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
