 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ker: The crystal structure of dTDP-D-glucose 4,6-dehydratase (RmlB) fr... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ker | ||||||
|---|---|---|---|---|---|---|---|
| Title | The crystal structure of dTDP-D-glucose 4,6-dehydratase (RmlB) from Streptococcus suis with dTDP-D-glucose bound | ||||||
 Components Components | dTDP-D-glucose 4,6-dehydratase | ||||||
 Keywords Keywords | LYASE / Rossmann fold | ||||||
| Function / homology |  Function and homology information Function and homology informationdTDP-glucose 4,6-dehydratase / dTDP-glucose 4,6-dehydratase activity / nucleotide-sugar metabolic process / dTDP-rhamnose biosynthetic process / lipopolysaccharide biosynthetic process / polysaccharide biosynthetic process / nucleotide binding Similarity search - Function | ||||||
| Biological species |  Streptococcus suis (bacteria) Streptococcus suis (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2.2 Å | ||||||
 Authors Authors | Allard, S.T.M. / Beis, K. / Giraud, M.-F. / Hegeman, A.D. / Gross, J.W. / Whitfield, C. / Graninger, M. / Messner, P. / Allen, A.G. / Naismith, J.H. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Toward a structural understanding of the dehydratase mechanism. Authors: Allard, S.T. / Beis, K. / Giraud, M.F. / Hegeman, A.D. / Gross, J.W. / Wilmouth, R.C. / Whitfield, C. / Graninger, M. / Messner, P. / Allen, A.G. / Maskell, D.J. / Naismith, J.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ker.cif.gz | 170.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ker.ent.gz | 132.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1ker.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1ker_validation.pdf.gz | 686.8 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1ker_full_validation.pdf.gz | 697.3 KB | Display | |
| Data in XML | 1ker_validation.xml.gz | 16.8 KB | Display | |
| Data in CIF | 1ker_validation.cif.gz | 28.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ke/1kerftp://data.pdbj.org/pub/pdb/validation_reports/ke/1ker | HTTPS FTP |
-Related structure data
| Related structure data |  1kepC  1ketC  1keuC  1kewC  1g1aS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 38982.277 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Streptococcus suis (bacteria) / Gene: rmlB / Variant: serotype 2 / Plasmid: pET21 / Species (production host): Escherichia coli / Production host: References: UniProt: P95780, UniProt: Q8GIP9*PLUS, dTDP-glucose 4,6-dehydratase #2: Chemical |   Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 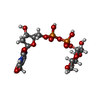  Mass: 564.329 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H26N2O16P2 Mass: 564.329 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C16H26N2O16P2#4: Chemical |   Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM Mass: 663.425 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C21H27N7O14P2 / Comment: NAD*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 712 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 712 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.45 Å3/Da / Density % sol: 64.36 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, hanging drop / pH: 5.4 Details: 30% w/v PEG 8000, 0.1M sodium cacodylate pH 5.4, 0.2M ammonium sulphate, VAPOR DIFFUSION, HANGING DROP at 293K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, sitting drop / Details: used microseeding | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: BM14 / Wavelength: 0.933 Å / Beamline: BM14 / Wavelength: 0.933 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jun 6, 2001 / Details: mirrors |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.933 Å / Relative weight: 1 |
| Reflection | Resolution: 2.2→49.7 Å / Num. obs: 55517 / % possible obs: 99.8 % / Redundancy: 3.2 % / Biso Wilson estimate: 19.232 Å2 / Rmerge(I) obs: 0.134 / Rsym value: 0.112 / Net I/σ(I): 5.7 |
| Reflection shell | Resolution: 2.2→2.32 Å / Redundancy: 3.3 % / Rmerge(I) obs: 0.34 / Mean I/σ(I) obs: 2.6 / Num. unique all: 8000 / Rsym value: 0.284 / % possible all: 99.9 |
| Reflection | *PLUS Num. measured all: 179806 / Rmerge(I) obs: 0.112 |
| Reflection shell | *PLUS % possible obs: 99.9 % / Num. unique obs: 8000 / Num. measured obs: 26125 / Rmerge(I) obs: 0.284 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB 1G1A Resolution: 2.2→49.7 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 17.17 Å2 | ||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.2→49.7 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| LS refinement shell | Resolution: 2.2→2.28 Å
| ||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.2 Å / Lowest resolution: 49.7 Å / % reflection Rfree: 10 % / Rfactor obs: 0.175 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.218 / Rfactor Rwork: 0.189 |
