 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1kc3: Crystal structure of dTDP-6-deoxy-L-lyxo-4-hexulose reductase (Rm... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1kc3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystal structure of dTDP-6-deoxy-L-lyxo-4-hexulose reductase (RmlD) in complex with NADPH and dTDP-L-rhamnose | ||||||
 Components Components | dTDP-glucose oxidoreductase | ||||||
 Keywords Keywords | OXIDOREDUCTASE / Rossman-fold / sugar-nucleotide-binding domain | ||||||
| Function / homology |  Function and homology information Function and homology informationdTDP-4-dehydrorhamnose reductase / dTDP-4-dehydrorhamnose reductase activity / O antigen biosynthetic process / dTDP-rhamnose biosynthetic process / lipopolysaccharide biosynthetic process / polysaccharide biosynthetic process / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  Salmonella typhimurium (bacteria) Salmonella typhimurium (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å | ||||||
 Authors Authors | Blankenfeldt, W. / Kerr, I.D. / Giraud, M.F. / McMiken, H.J. / Leonard, G.A. / Whitfield, C. / Messner, P. / Graninger, M. / Naismith, J.H. | ||||||
 Citation Citation | Journal: Structure / Year: 2002 Title: Variation on a theme of SDR. dTDP-6-deoxy-L- lyxo-4-hexulose reductase (RmlD) shows a new Mg2+-dependent dimerization mode. Authors: Blankenfeldt, W. / Kerr, I.D. / Giraud, M.F. / McMiken, H.J. / Leonard, G. / Whitfield, C. / Messner, P. / Graninger, M. / Naismith, J.H. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1999Title: Overexpression, purification, crystallisation and preliminary structural study of dTDP-6-deoxy-lyxo-4-reductase (RmlD) the fourth enzyme of the dTDP-rhanose synthesis pathway,from Salmonella ...Title: Overexpression, purification, crystallisation and preliminary structural study of dTDP-6-deoxy-lyxo-4-reductase (RmlD) the fourth enzyme of the dTDP-rhanose synthesis pathway,from Salmonella entrica serovar Typhimurim Authors: Giraud, M.F. / McMiken, H.J. / Leonard, G.A. / Messner, P. / Whitfield, C. / Naismith, J.H. #2: Journal: Curr.Opin.Struct.Biol. / Year: 2000Title: The rhamnose pathway Authors: Giraud, M.F. / Naismith, J.H. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1kc3.cif.gz | 77.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1kc3.ent.gz | 56.6 KB | Display | PDB format |
| PDBx/mmJSON format | 1kc3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/kc/1kc3ftp://data.pdbj.org/pub/pdb/validation_reports/kc/1kc3 | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||||
| Unit cell |
| ||||||||||
| Components on special symmetry positions |
| ||||||||||
| Details | The biological assembly is a dimer generated by the twofold axis along c: -x, -y, z. |
-Components
| #1: Protein | Mass: 32589.943 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Salmonella typhimurium (bacteria) / Gene: rfbA / Plasmid: pET30 / Production host: References: UniProt: P26392, dTDP-4-dehydrorhamnose reductase |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #3: Chemical | ChemComp-NDP / 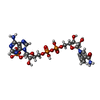  Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 Mass: 745.421 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H30N7O17P3 |
| #4: Chemical | ChemComp-TRH / 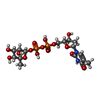  Mass: 548.330 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H26N2O15P2 Mass: 548.330 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C16H26N2O15P2 |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 93 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 44.04 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop / pH: 6.8 Details: HEPES, PEG-MME 5000, MgCl2, pH 6.8, VAPOR DIFFUSION, SITTING DROP at 298K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.1 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 Å / Beamline: ID14-1 / Wavelength: 0.934 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Aug 27, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→28.75 Å / Num. all: 7905 / Num. obs: 7905 / % possible obs: 99.1 % / Observed criterion σ(I): 1.8 / Redundancy: 3.5 % / Biso Wilson estimate: 66 Å2 / Rmerge(I) obs: 0.105 / Net I/σ(I): 3.8 |
| Reflection shell | Resolution: 2.7→2.77 Å / Redundancy: 3.4 % / Rmerge(I) obs: 0.377 / Mean I/σ(I) obs: 1.8 / % possible all: 99.3 |
| Reflection | *PLUS Num. measured all: 28521 / Rmerge(I) obs: 0.105 |
| Reflection shell | *PLUS % possible obs: 99.3 % / Rmerge(I) obs: 0.377 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.7→28.87 Å / SU B: 18.382 / SU ML: 0.364 / SU Rfree: 0.449 / Isotropic thermal model: isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R Free: 0.449 / Stereochemistry target values: Engh & Huber Details: TLS-option with cofactor-binding, substrate-binding domains and waters as three separate TLS-bodies was used throughout
| ||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 39.02 Å2
| ||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error free: 0.449 Å | ||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→28.87 Å
| ||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.7→2.77 Å / Total num. of bins used: 20 /
| ||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor all: 0.198697 / Rfactor obs: 0.196 / Rfactor Rfree: 0.289 / Rfactor Rwork: 0.191 | ||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.291 / Rfactor Rwork: 0.213 |
