 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1k1k: Structure of Mutant Human Carbonmonoxyhemoglobin C (beta E6K) at ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1k1k | ||||||
|---|---|---|---|---|---|---|---|
| Title | Structure of Mutant Human Carbonmonoxyhemoglobin C (beta E6K) at 2.0 Angstrom Resolution in Phosphate Buffer. | ||||||
 Components Components |
| ||||||
 Keywords Keywords | OXYGEN STORAGE/TRANSPORT / mutant human hemoglobin C(betaE6K) / OXYGEN STORAGE-TRANSPORT COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology informationcellular oxidant detoxification / Heme assimilation / nitric oxide transport / hemoglobin alpha binding / hemoglobin binding / haptoglobin-hemoglobin complex / renal absorption / hemoglobin complex / oxygen transport / Scavenging of heme from plasma ...cellular oxidant detoxification / Heme assimilation / nitric oxide transport / hemoglobin alpha binding / hemoglobin binding / haptoglobin-hemoglobin complex / renal absorption / hemoglobin complex / oxygen transport / Scavenging of heme from plasma / erythrocyte development / endocytic vesicle lumen / blood vessel diameter maintenance / hydrogen peroxide catabolic process / oxygen carrier activity / carbon dioxide transport / response to hydrogen peroxide / Heme signaling / Erythrocytes take up oxygen and release carbon dioxide / Erythrocytes take up carbon dioxide and release oxygen / Cytoprotection by HMOX1 / oxygen binding / Late endosomal microautophagy / platelet aggregation / regulation of blood pressure / Chaperone Mediated Autophagy / positive regulation of nitric oxide biosynthetic process / tertiary granule lumen / Factors involved in megakaryocyte development and platelet production / blood microparticle / ficolin-1-rich granule lumen / iron ion binding / inflammatory response / heme binding / Neutrophil degranulation / : / extracellular exosome / extracellular region / membrane / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Dewan, J.C. / Taylor-Feeling, A. / Puius, Y.A. / Patskovska, L. / Patskovsky, Y. / Nagel, R.L. / Almo, S.C. / Hirsch, R.E. | ||||||
 Citation Citation | Journal: Acta Crystallogr.,Sect.D / Year: 2002 Title: Structure of mutant human carbonmonoxyhemoglobin C (betaE6K) at 2.0 A resolution. Authors: Dewan, J.C. / Feeling-Taylor, A. / Puius, Y.A. / Patskovska, L. / Patskovsky, Y. / Nagel, R.L. / Almo, S.C. / Hirsch, R.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1k1k.cif.gz | 72.9 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1k1k.ent.gz | 53.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1k1k.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/k1/1k1kftp://data.pdbj.org/pub/pdb/validation_reports/k1/1k1k | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1hhoS S: Starting model for refinement |
|---|---|
| Similar structure data |





















-Links
 PDBj
PDBj
















- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | The biological assembly is a tetramer generated from the dimer in the assymetric unit by the two fold axis: -y,-x,1/2-z. |
-Components
| #1: Protein | Mass: 15150.353 Da / Num. of mol.: 1 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / Cell: erythrocyte / Tissue: BLOOD / References: UniProt: P69905 | ||||
|---|---|---|---|---|---|
| #2: Protein | Mass: 15890.265 Da / Num. of mol.: 1 / Mutation: E6K / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / Cell: erythrocyte / Tissue: BLOOD / References: UniProt: P68871 | ||||
| #3: Chemical |   Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4 Mass: 616.487 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C34H32FeN4O4#4: Chemical | 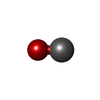  Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO Mass: 28.010 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: CO#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 113 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 113 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.66 % | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 310 K / Method: batch method / pH: 7.35 Details: potassium phosphate 1.75M, pH 7.35, batch method, temperature 310K | ||||||||||||||||||
| Crystal grow | *PLUS | ||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 Å |
| Detector | Type: SIEMENS X1000 / Detector: AREA DETECTOR / Date: Mar 8, 1995 |
| Radiation | Monochromator: graphite / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2→22.55 Å / Num. all: 19382 / Num. obs: 19382 / % possible obs: 94 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.38 % / Biso Wilson estimate: 24.9 Å2 / Rmerge(I) obs: 0.063 |
| Reflection shell | Resolution: 2→2.09 Å / Num. unique all: 2135 / % possible all: 85.16 |
| Reflection | *PLUS Highest resolution: 2 Å / Lowest resolution: 22.5 Å / % possible obs: 94 % / Num. measured all: 84880 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1HHO Resolution: 2→22.55 Å / Isotropic thermal model: Isotropic / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 Details: The side chain of beta K6 was added to the model in weak continuous density present at the 0.5 sigma level in a 2Fo-Fc map.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24.9 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→22.55 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Refine-ID: X-RAY DIFFRACTION / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 22.5 Å / Rfactor Rfree: 0.238 / Rfactor Rwork: 0.183 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
