 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1joh | ||||||
|---|---|---|---|---|---|---|---|
| Title | THE STRUCTURE OF ANTIAMOEBIN I, A MEMBRANE-ACTIVE PEPTIDE | ||||||
 Components Components | ANTIAMOEBIN I | ||||||
 Keywords Keywords | ANTIBIOTIC / ANTIAMOEBIN I / PEPTAIBOL / ANTIBACTERIAL / ANTIFUNGAL | ||||||
| Function / homology | Antiamoebin 1 / METHANOL / :  Function and homology information Function and homology information | ||||||
| Biological species |  EMERICELLOPSIS (fungus) EMERICELLOPSIS (fungus) | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.4 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.4 Å | ||||||
 Authors Authors | Snook, C.F. / Wallace, B.A. | ||||||
 Citation Citation | Journal: Structure / Year: 1998 Title: The Structure and Function of Antiamoebin I, a Proline-Rich Membrane-Active Polypeptide. Authors: Snook, C.F. / Woolley, G.A. / Oliva, G. / Pattabhi, V. / Wood, S.F. / Blundell, T.L. / Wallace, B.A. #1: Journal: Acta Crystallogr.,Sect.D / Year: 1999 Title: The Molecular-Replacement Solution of an Intermediate-Sized Helical Polypeptide, Antiamoebin I. Authors: Snook, C.F. / Wallace, B.A. #2: Journal: Thesis, University of London / Year: 1997Title: The Structure and Function of Antiamoebin I, a Membrane-Active Peptide Authors: Snook, C.F. #3: Journal: Thesis, University of London / Year: 1988Title: Molecular Redundancy and Protein Crystallography : X-Ray Structure Analysis of Antiamoebin I, Bovine Pancreatic Polypeptide and Human Serum Amyloid P Component Authors: Oliva, G. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1joh.cif.gz | 18.5 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1joh.ent.gz | 12.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1joh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jo/1johftp://data.pdbj.org/pub/pdb/validation_reports/jo/1joh | HTTPS FTP |
|---|
-Related structure data
| Related structure data | |
|---|---|
| Similar structure data |













-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 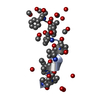
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given / Matrix: (1), |
-Components
| #1: Protein/peptide |   Type: Peptaibol / Class: Antibiotic / Mass: 1654.991 Da / Num. of mol.: 2 / Source method: isolated from a natural source Type: Peptaibol / Class: Antibiotic / Mass: 1654.991 Da / Num. of mol.: 2 / Source method: isolated from a natural sourceDetails: ANTIAMOEBIN I IS A HEXADECAMERIC HELICAL PEPTIDE. THE N-TERM IS ACETYLATED (RESIDUE 0) Source: (natural) EMERICELLOPSIS (fungus) / References: NOR: NOR00945, Antiamoebin 1#2: Chemical | ChemComp-MOH /   Mass: 32.042 Da / Num. of mol.: 23 / Source method: obtained synthetically / Formula: CH4O Mass: 32.042 Da / Num. of mol.: 23 / Source method: obtained synthetically / Formula: CH4OCompound details | ANTIAMOEBIN I IS LINEAR PEPTIDE, A MEMBER OF THE PEPTAIBOL FAMILY OF MEMBRANE CHANNEL FORMING ...ANTIAMOEBI | Has protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 1.71 Å3/Da / Density % sol: 28 % / Description: USED NORMALISED STRUCTURE FACTORS |
|---|---|
| Crystal grow | *PLUS Method: unknown |
| Components of the solutions | *PLUS Conc.: 170 mg/ml / Common name: peptide |
-Data collection
| Diffraction | Mean temperature: 293 K |
|---|---|
| Diffraction source | Source: SEALED TUBE / Wavelength: 1.5418 |
| Detector | Type: HILGER-WATTS / Detector: DIFFRACTOMETER / Date: 1986 |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.2→25 Å / Num. obs: 6715 / % possible obs: 93.7 % / Observed criterion σ(I): 0 / Redundancy: 1 % / Rmerge(I) obs: 0.012 / Net I/σ(I): 10.5 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: RESIDUES 6 - 16 OF LEU1-ZERVAMICIN WITH ALL NON -EQUIVALENT SIDE-CHAINS TRIMMED TO ALA. Resolution: 1.4→25 Å / Num. parameters: 1074 / Num. restraintsaints: 982 / σ(F): 0 / StereochEM target val spec case: HETATM DATA FROM CCSD / Stereochemistry target values: ENGH AND HUBER Details: REFINEMENT STARTED USING 10.0 TO 2.5A AND DATA CUT-OFF AT 2 SIGMA IN X-PLOR. HIGH RESOLUTION LIMIT INCREASED TO 2.0A, LOW RESOLUTION DECREASED TO 25.0A AND DATA CUT-OFF DECREASED TO 0.0 ...Details: REFINEMENT STARTED USING 10.0 TO 2.5A AND DATA CUT-OFF AT 2 SIGMA IN X-PLOR. HIGH RESOLUTION LIMIT INCREASED TO 2.0A, LOW RESOLUTION DECREASED TO 25.0A AND DATA CUT-OFF DECREASED TO 0.0 SIGMA AT 2.0A RESOLUTION. RESOLUTION INCREASED FROM 2.0A TO 1.4A WITH ALL DATA USING SHELXL-93. REFINEMENT CEASED AFTER RESIDUES 1 TO 5 IN B-CHAIN IDENTIFIED AS DISORDERED IN DENSITY.
| |||||||||||||||||||||||||||||||||
| Refine analyze | Occupancy sum non hydrogen: 268 | |||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.4→25 Å
| |||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor all: 0.156 | |||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
