 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1jje: IMP-1 METALLO BETA-LACTAMASE FROM PSEUDOMONAS AERUGINOSA IN COMPL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1jje | ||||||
|---|---|---|---|---|---|---|---|
| Title | IMP-1 METALLO BETA-LACTAMASE FROM PSEUDOMONAS AERUGINOSA IN COMPLEX WITH A BIARYL SUCCINIC ACID INHIBITOR (11) | ||||||
 Components Components | IMP-1 METALLO BETA-LACTAMASE | ||||||
 Keywords Keywords | HYDROLASE / METALLO-BETA-LACTAMASE INHIBITOR / SUCCINIC ACID INHIBITOR / IMP-1 METALLO-BETA-LACTAMASE | ||||||
| Function / homology |  Function and homology information Function and homology informationHydrolases; Acting on ester bonds; Endoribonucleases producing 5'-phosphomonoesters / antibiotic catabolic process / beta-lactamase activity / beta-lactamase / periplasmic space / response to antibiotic / zinc ion binding Similarity search - Function | ||||||
| Biological species |   Pseudomonas aeruginosa (bacteria) Pseudomonas aeruginosa (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.8 Å X-RAY DIFFRACTION / SYNCHROTRON / FOURIER SYNTHESIS / Resolution: 1.8 Å | ||||||
 Authors Authors | Fitzgerald, P.M.D. / Sharma, N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Succinic acids as potent inhibitors of plasmid-borne IMP-1 metallo-beta-lactamase. Authors: Toney, J.H. / Hammond, G.G. / Fitzgerald, P.M. / Sharma, N. / Balkovec, J.M. / Rouen, G.P. / Olson, S.H. / Hammond, M.L. / Greenlee, M.L. / Gao, Y.D. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1jje.cif.gz | 104.6 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1jje.ent.gz | 79.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1jje.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1jje_validation.pdf.gz | 1.1 MB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1jje_full_validation.pdf.gz | 1.1 MB | Display | |
| Data in XML | 1jje_validation.xml.gz | 12.4 KB | Display | |
| Data in CIF | 1jje_validation.cif.gz | 18.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/jj/1jjeftp://data.pdbj.org/pub/pdb/validation_reports/jj/1jje | HTTPS FTP |
-Related structure data











-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24535.006 Da / Num. of mol.: 2 / Fragment: metallo-beta-lactamase Source method: isolated from a genetically manipulated source Details: PLEASE NOTE THAT THE PROTEIN CONTAINS POSTRANSLATIONAL MODIFICATION: ATOM CG OF ASN 26 IS BOUND TO ATOM N OF GLY 27. THIS ACCOUNTS FOR MISSING ATOMS LISTED IN REMARK 470 AND DISTORTED ...Details: PLEASE NOTE THAT THE PROTEIN CONTAINS POSTRANSLATIONAL MODIFICATION: ATOM CG OF ASN 26 IS BOUND TO ATOM N OF GLY 27. THIS ACCOUNTS FOR MISSING ATOMS LISTED IN REMARK 470 AND DISTORTED GEOMETRY REPORTED FOR THESE RESIDUES. Source: (gene. exp.) Pseudomonas aeruginosa (bacteria) / Production host: References: UniProt: P52699, UniProt: Q79MP6*PLUS, beta-lactamase #2: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 6 / Source method: obtained synthetically / Formula: Zn#3: Chemical |   Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2 Mass: 59.044 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H3O2#4: Chemical | 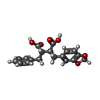  Mass: 342.343 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H18O6 Mass: 342.343 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C19H18O6#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 184 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.98 Å3/Da / Density % sol: 38.3 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 310 K / Method: vapor diffusion, hanging drop / pH: 6.9 Details: PEG MONOMETHYL ETHER 550, ZINC ACETATE, MES PH 6.7-7.1, 310 K, pH 6.9, VAPOR DIFFUSION, HANGING DROP, temperature 310K | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 37 ℃ / pH: 7.2 | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 / Wavelength: 1 Å | |||||||||
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Jun 22, 1999 | |||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||
| Radiation wavelength |
| |||||||||
| Reflection | Resolution: 1.8→99 Å / Num. all: 35791 / Num. obs: 35791 / % possible obs: 99.7 % / Observed criterion σ(F): -3 / Observed criterion σ(I): -3 / Redundancy: 3.59 % / Biso Wilson estimate: 52.2 Å2 / Rmerge(I) obs: 0.0752 / Net I/σ(I): 13.005 | |||||||||
| Reflection shell | Resolution: 1.8→1.91 Å / Redundancy: 3.7 % / Rmerge(I) obs: 0.4997 / Mean I/σ(I) obs: 1.336 / Num. unique all: 5910 / % possible all: 100 | |||||||||
| Reflection | *PLUS Lowest resolution: 9999 Å | |||||||||
| Reflection shell | *PLUS % possible obs: 100 % / Num. unique obs: 5910 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: FOURIER SYNTHESIS Starting model: ANOTHER INHIBITED COMPLEX IN THE SAME SPACE GROUP Resolution: 1.8→10 Å / Num. parameters: 14767 / Num. restraintsaints: 14533 / Cross valid method: THROUGHOUT / σ(F): -3 / σ(I): -3 / Stereochemistry target values: Engh & Huber
| |||||||||||||||||||||||||||
| Solvent computation | Solvent model: SHELXL 97 SWAT PARAMETER REFINEMENT | |||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 35.2 Å2 | |||||||||||||||||||||||||||
| Refine analyze | Num. disordered residues: 0 / Occupancy sum hydrogen: 0 / Occupancy sum non hydrogen: 3688 | |||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.8→10 Å
| |||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): -3 / % reflection Rfree: 5 % | |||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_plane_restr / Dev ideal: 0.337 | |||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.8 Å / Lowest resolution: 1.87 Å / Num. reflection obs: 3653 / Rfactor obs: 0.365 |
