 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1j78 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Crystallographic analysis of the human vitamin D binding protein | ||||||
 Components Components | vitamin D binding protein | ||||||
 Keywords Keywords | TRANSPORT / LIGAND BINDING PROTEIN / plasma protein / vitamin D binding / actin binding / fatty acid binding / GC-globulin / Group-specific component | ||||||
| Function / homology |  Function and homology information Function and homology informationcalcidiol binding / vitamin transmembrane transporter activity / vitamin transport / Vitamin D (calciferol) metabolism / vitamin D metabolic process / vitamin D binding / lysosomal lumen / response to nutrient levels / actin binding / blood microparticle ...calcidiol binding / vitamin transmembrane transporter activity / vitamin transport / Vitamin D (calciferol) metabolism / vitamin D metabolic process / vitamin D binding / lysosomal lumen / response to nutrient levels / actin binding / blood microparticle / : / extracellular exosome / extracellular region / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / COMBINED MAD, MIRAS / Resolution: 2.31 Å X-RAY DIFFRACTION / SYNCHROTRON / COMBINED MAD, MIRAS / Resolution: 2.31 Å | ||||||
 Authors Authors | Verboven, C. / Rabijns, A. / De Maeyer, M. / Van Baelen, H. / Bouillon, R. / De Ranter, C. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 2002 Title: A structural basis for the unique binding features of the human vitamin D-binding protein. Authors: Verboven, C. / Rabijns, A. / De Maeyer, M. / Van Baelen, H. / Bouillon, R. / De Ranter, C. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1j78.cif.gz | 187.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1j78.ent.gz | 149.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1j78.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/j7/1j78ftp://data.pdbj.org/pub/pdb/validation_reports/j7/1j78 | HTTPS FTP |
|---|
-Related structure data











-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 51277.289 Da / Num. of mol.: 2 / Source method: isolated from a natural source / Source: (natural) Homo sapiens (human) / References: UniProt: P02774#2: Chemical | 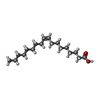  Mass: 282.461 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C18H34O2 Mass: 282.461 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C18H34O2#3: Chemical | ChemComp-VDY / | 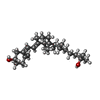  Mass: 400.637 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H44O2 Mass: 400.637 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H44O2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 299 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.12 Å3/Da / Density % sol: 60.56 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 293 K / Method: vapor diffusion, sitting drop / pH: 4.6 Details: PEG 400, sodium acetate, pH 4.6, VAPOR DIFFUSION, SITTING DROP, temperature 20K | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging dropDetails: Verboven, C.C., (1995) J. Steroid Biochem. Mol. Biol., 54, 11. | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: X11 / Wavelength: 0.9116 Å / Beamline: X11 / Wavelength: 0.9116 Å |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 17, 1999 / Details: Bent mirror |
| Radiation | Monochromator: Triangular monochromator / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9116 Å / Relative weight: 1 |
| Reflection | Resolution: 2.31→30 Å / Num. all: 55933 / Num. obs: 55444 / % possible obs: 99.2 % / Observed criterion σ(I): -3 / Redundancy: 3.4 % / Biso Wilson estimate: 50.6 Å2 / Rmerge(I) obs: 0.031 / Rsym value: 0.031 / Net I/σ(I): 22.7 |
| Reflection shell | Resolution: 2.31→2.35 Å / Redundancy: 3.4 % / Rmerge(I) obs: 0.209 / Mean I/σ(I) obs: 5.2 / Num. unique all: 2766 / Rsym value: 0.209 / % possible all: 99.6 |
| Reflection | *PLUS Lowest resolution: 30 Å / Num. measured all: 187531 |
| Reflection shell | *PLUS % possible obs: 99.6 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: COMBINED MAD, MIRAS / Resolution: 2.31→29.38 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 1255175.19 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / Stereochemistry target values: Engh & Huber Details: There are two molecules in the asymmetric unit: chain A and B. For chain A residues 1-12, 60-67, 99-102 and 458 and for chain B residues 1-2, 98-104 and 457-458 are disordered and have not ...Details: There are two molecules in the asymmetric unit: chain A and B. For chain A residues 1-12, 60-67, 99-102 and 458 and for chain B residues 1-2, 98-104 and 457-458 are disordered and have not been included in the model.
| ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 45.26 Å2 / ksol: 0.307 e/Å3 | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 60.5 Å2
| ||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.31→29.38 Å
| ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.31→2.42 Å / Rfactor Rfree error: 0.02 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Num. reflection obs: 53194 / σ(F): 0 / Num. reflection Rfree: 2236 / % reflection Rfree: 4 % / Rfactor obs: 0.2201 / Rfactor Rfree: 0.2518 / Rfactor Rwork: 0.22 | ||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 60.5 Å2 | ||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.351 / % reflection Rfree: 4.5 % / Rfactor Rwork: 0.29 |
