解像度: 2.31→2.35 Å / 冗長度: 3.4 % / Rmerge(I) obs: 0.209 / Mean I/σ(I) obs: 5.2 / Num. unique all: 2766 / Rsym value: 0.209 / % possible all: 99.6
反射
*PLUS
最低解像度: 30 Å / Num. measured all: 187531
反射 シェル
*PLUS
% possible obs: 99.6 %
-
解析
ソフトウェア
名称
バージョン
分類
DENZO
データ削減
SCALEPACK
データスケーリング
SHARP
位相決定
CNS
1
精密化
精密化
構造決定の手法: COMBINED MAD, 多重同系置換・異常分散 解像度: 2.31→29.38 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 1255175.19 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / 立体化学のターゲット値: Engh & Huber 詳細: There are two molecules in the asymmetric unit: chain A and B. For chain A residues 1-12, 60-67, 99-102 and 458 and for chain B residues 1-2, 98-104 and 457-458 are disordered and have not ...詳細: There are two molecules in the asymmetric unit: chain A and B. For chain A residues 1-12, 60-67, 99-102 and 458 and for chain B residues 1-2, 98-104 and 457-458 are disordered and have not been included in the model.
 ムービー
ムービー コントローラー
コントローラー


 データを開く
データを開く
 基本情報
基本情報 要素
要素 キーワード
キーワード LIGAND BINDING PROTEIN (リガンド) /
LIGAND BINDING PROTEIN (リガンド) /  機能・相同性情報
機能・相同性情報
 データ登録者
データ登録者 引用
引用 構造の表示
構造の表示 ダウンロードとリンク
ダウンロードとリンク その他のダウンロード
その他のダウンロード










 PDBj
PDBj







 集合体
集合体


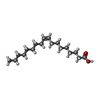
 分子量: 282.461 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C18H34O2
分子量: 282.461 Da / 分子数: 3 / 由来タイプ: 合成 / 式: C18H34O2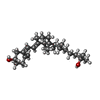
 分子量: 400.637 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C27H44O2
分子量: 400.637 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C27H44O2 分子量: 18.015 Da / 分子数: 299 / 由来タイプ: 天然 / 式: H2O
分子量: 18.015 Da / 分子数: 299 / 由来タイプ: 天然 / 式: H2O 試料調製
試料調製 / ビームライン: X11 / 波長: 0.9116 Å
/ ビームライン: X11 / 波長: 0.9116 Å 解析
解析