 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1i9j: TESTOSTERONE COMPLEX STRUCTURE OF THE RECOMBINANT MONOCLONAL WILD... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1i9j | ||||||
|---|---|---|---|---|---|---|---|
| Title | TESTOSTERONE COMPLEX STRUCTURE OF THE RECOMBINANT MONOCLONAL WILD TYPE ANTI-TESTOSTERONE FAB FRAGMENT | ||||||
 Components Components |
| ||||||
 Keywords Keywords | IMMUNE SYSTEM / fab fragment / anti-testosterone / recombinant / monoclonal / testosterone | ||||||
| Function / homology |  Function and homology information Function and homology informationimmunoglobulin complex / adaptive immune response / extracellular region / metal ion binding / plasma membrane Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Valjakka, J. / Takkinenz, K. / Teerinen, T. / Soderlund, H. / Rouvinen, J. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2002 Title: Structural insights into steroid hormone binding: the crystal structure of a recombinant anti-testosterone Fab fragment in free and testosterone-bound forms. Authors: Valjakka, J. / Takkinenz, K. / Teerinen, T. / Soderlund, H. / Rouvinen, J. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2000Title: X-ray studies of recombinant anti-testosterone fab fragments: the use of peg 3350 in crystallization Authors: Valjakka, J. / Hemminki, A. / Teerinen, T. / Takkinen, K. / Rouvinen, J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1i9j.cif.gz | 104.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1i9j.ent.gz | 79.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1i9j.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i9/1i9jftp://data.pdbj.org/pub/pdb/validation_reports/i9/1i9j | HTTPS FTP |
|---|
-Related structure data





















-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Antibody | Mass: 24056.625 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Antibody | Mass: 23509.566 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) |
| #3: Chemical | ChemComp-TES / 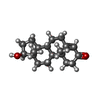  Mass: 288.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H28O2 / Comment: hormone*YM Mass: 288.424 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C19H28O2 / Comment: hormone*YM |
| #4: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 337 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 337 / Source method: isolated from a natural source / Formula: H2O |
| Has protein modification | Y |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.03 Å3/Da / Density % sol: 59.44 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, hanging drop Details: peg3350, VAPOR DIFFUSION, HANGING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 5.6 | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 120 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200HB / Wavelength: 1.5418 Å |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Details: MSC Confocal Blue Optics |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→99 Å / Num. all: 52788 / Num. obs: 16913 / % possible obs: 91.7 % / Observed criterion σ(I): 1 / Redundancy: 3.1 % / Rmerge(I) obs: 0.085 |
| Reflection shell | Resolution: 2.6→2.69 Å / Rmerge(I) obs: 0.299 / % possible all: 95.7 |
| Reflection | *PLUS Lowest resolution: 99 Å / Num. measured all: 52788 |
| Reflection shell | *PLUS % possible obs: 95.7 % / Mean I/σ(I) obs: 4.2 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: native anti-testosterone fab fragment Resolution: 2.6→99 Å / σ(F): 1
| ||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→99 Å
| ||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 500 Å / σ(F): 1 / Rfactor obs: 0.188 | ||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
