 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1i70 | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF RNASE SA Y86F MUTANT | ||||||
 要素 要素 | GUANYL-SPECIFIC RIBONUCLEASE SA | ||||||
 キーワード キーワード | HYDROLASE / MUTANT | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報ribonuclease T1 / ribonuclease T1 activity / RNA endonuclease activity / lyase activity / RNA binding / extracellular region 類似検索 - 分子機能 | ||||||
| 生物種 |  Streptomyces aureofaciens (バクテリア) Streptomyces aureofaciens (バクテリア) | ||||||
| 手法 |  X線回折 / シンクロトロン / STARTING MODEL WAS USED WITHOUT MOLECULAR REPLACEMENT / 解像度: 1.7 Å X線回折 / シンクロトロン / STARTING MODEL WAS USED WITHOUT MOLECULAR REPLACEMENT / 解像度: 1.7 Å | ||||||
 データ登録者 データ登録者 | Sevcik, J. / Urbanikova, L. | ||||||
 引用 引用 | ジャーナル: J.Mol.Biol. / 年: 2001 タイトル: Tyrosine hydrogen bonds make a large contribution to protein stability. 著者: Pace, C.N. / Horn, G. / Hebert, E.J. / Bechert, J. / Shaw, K. / Urbanikova, L. / Scholtz, J.M. / Sevcik, J. | ||||||
| 履歴 |
| ||||||
| Remark 999 | SEQUENCE ACCORDING TO THE AUTHOR, THE ORIGINAL SEQUENCE OF THE RNASE SA IN THE SEQUENCE DATABASE ...SEQUENCE ACCORDING TO THE AUTHOR, THE ORIGINAL SEQUENCE OF THE RNASE SA IN THE SEQUENCE DATABASE SWISSPROT ENTRY P05798 IS WRONG. RESIDUE 72 IS THR, NOT CYS. THIS ISSUE IS DISCUSSED IN THE PAPER: SEVCIK J., DAUTER Z., LAMZIN V.S. AND WILSON K.S. (1996), ACTA CRYSTALLOGR. D52, 327-344 ON THE BASIS OF ATOMIC RESOLUTION REFINEMENT. |
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1i70.cif.gz | 56.8 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1i70.ent.gz | 41.1 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1i70.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| 文書・要旨 | 1i70_validation.pdf.gz | 425.4 KB | 表示 | wwPDB検証レポート |
|---|---|---|---|---|
| 文書・詳細版 | 1i70_full_validation.pdf.gz | 431.1 KB | 表示 | |
| XML形式データ | 1i70_validation.xml.gz | 13.6 KB | 表示 | |
| CIF形式データ | 1i70_validation.cif.gz | 20.1 KB | 表示 | |
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/i7/1i70ftp://data.pdbj.org/pub/pdb/validation_reports/i7/1i70 | HTTPS FTP |
-関連構造データ





-リンク
 PDBj
PDBj- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 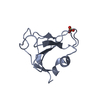
| ||||||||
| 2 | 
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 10566.492 Da / 分子数: 2 / 変異: Y86F / 由来タイプ: 組換発現 由来: (組換発現) Streptomyces aureofaciens (バクテリア)発現宿主: #2: 化合物 | ChemComp-SO4 / |   分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4 分子量: 96.063 Da / 分子数: 1 / 由来タイプ: 合成 / 式: SO4#3: 水 | ChemComp-HOH / |  分子量: 18.015 Da / 分子数: 305 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 305 / 由来タイプ: 天然 / 式: H2OHas protein modification | Y | |
|---|
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.34 Å3/Da / 溶媒含有率: 47 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 293 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 7 詳細: AMMONIUM SULFATE, MONOSODIUM PHOSPHATE, DISODIUM PHOSPHATE, pH 7.0, VAPOR DIFFUSION, HANGING DROP, temperature 293K | ||||||||||||||||||||
| 結晶化 | *PLUS 詳細: used microseeding / PH range low: 7.2 / PH range high: 7 | ||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 293 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: LURE  / ビームライン: D41A / 波長: 1.38 Å / ビームライン: D41A / 波長: 1.38 Å |
| 検出器 | タイプ: MARRESEARCH / 検出器: IMAGE PLATE / 日付: 1999年7月22日 |
| 放射 | モノクロメーター: FOCUSING CURVED MONO. / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.38 Å / 相対比: 1 |
| 反射 | 解像度: 1.7→30.7 Å / Num. all: 21828 / Num. obs: 84702 / % possible obs: 97.1 % / Observed criterion σ(F): 0 / 冗長度: 3.9 % / Biso Wilson estimate: 13.9 Å2 / Rmerge(I) obs: 0.061 / Net I/σ(I): 20.8 |
| 反射 シェル | 解像度: 1.7→1.76 Å / Rmerge(I) obs: 0.218 / Mean I/σ(I) obs: 7.1 / % possible all: 95.5 |
| 反射 | *PLUS Num. obs: 21859 / Num. measured all: 84702 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: STARTING MODEL WAS USED WITHOUT MOLECULAR REPLACEMENT 開始モデル: PDB ENTRY 1RGG 解像度: 1.7→30.7 Å / σ(I): 0 / ESU R: 0.13 / ESU R Free: 0.09 / 立体化学のターゲット値: ENGH & HUBER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 16.85 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati d res low obs: 30.7 Å / Luzzati sigma a obs: 0.014 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.7→30.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 1.7→1.76 Å / % reflection obs: 95.5 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: REFMAC / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS 最高解像度: 1.7 Å / % reflection Rfree: 5 % / Rfactor Rwork: 0.13 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS タイプ: p_planar_d / Dev ideal: 0.04 / Dev ideal target: 0.05 |
