 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1i6i: CRYSTAL STRUCTURE OF THE KIF1A MOTOR DOMAIN COMPLEXED WITH MG-AMPPCP -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1i6i | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE KIF1A MOTOR DOMAIN COMPLEXED WITH MG-AMPPCP | ||||||
 Components Components | KINESIN-LIKE PROTEIN KIF1A | ||||||
 Keywords Keywords | TRANSPORT PROTEIN / kinesin / motor protein / catalytic core | ||||||
| Function / homology |  Function and homology information Function and homology informationanterograde dendritic transport / interkinetic nuclear migration / anterograde synaptic vesicle transport / protein transport along microtubule / neuronal dense core vesicle membrane / dense core granule cytoskeletal transport / plus-end-directed kinesin ATPase / Kinesins / anterograde neuronal dense core vesicle transport / retrograde neuronal dense core vesicle transport ...anterograde dendritic transport / interkinetic nuclear migration / anterograde synaptic vesicle transport / protein transport along microtubule / neuronal dense core vesicle membrane / dense core granule cytoskeletal transport / plus-end-directed kinesin ATPase / Kinesins / anterograde neuronal dense core vesicle transport / retrograde neuronal dense core vesicle transport / regulation of dendritic spine development / COPI-dependent Golgi-to-ER retrograde traffic / MHC class II antigen presentation / regulation of dendritic spine morphogenesis / anterograde axonal transport / plus-end-directed microtubule motor activity / kinesin complex / neuronal dense core vesicle / vesicle-mediated transport / axon cytoplasm / dendrite cytoplasm / synaptic vesicle / presynapse / microtubule binding / microtubule / neuron projection / postsynapse / axon / neuronal cell body / dendrite / perinuclear region of cytoplasm / glutamatergic synapse / ATP hydrolysis activity / protein-containing complex / ATP binding / metal ion binding / identical protein binding / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Kikkawa, M. / Sablin, E.P. / Okada, Y. / Yajima, H. / Fletterick, R.J. / Hirokawa, N. | ||||||
 Citation Citation | Journal: Nature / Year: 2001 Title: Switch-based mechanism of kinesin motors. Authors: M Kikkawa / E P Sablin / Y Okada / H Yajima / R J Fletterick / N Hirokawa /  Abstract: Kinesin motors are specialized enzymes that use hydrolysis of ATP to generate force and movement along their cellular tracks, the microtubules. Although numerous biochemical and biophysical studies ...Kinesin motors are specialized enzymes that use hydrolysis of ATP to generate force and movement along their cellular tracks, the microtubules. Although numerous biochemical and biophysical studies have accumulated much data that link microtubule-assisted ATP hydrolysis to kinesin motion, the structural view of kinesin movement remains unclear. This study of the monomeric kinesin motor KIF1A combines X-ray crystallography and cryo-electron microscopy, and allows analysis of force-generating conformational changes at atomic resolution. The motor is revealed in its two functionally critical states-complexed with ADP and with a non-hydrolysable analogue of ATP. The conformational change observed between the ADP-bound and the ATP-like structures of the KIF1A catalytic core is modular, extends to all kinesins and is similar to the conformational change used by myosin motors and G proteins. Docking of the ADP-bound and ATP-like crystallographic models of KIF1A into the corresponding cryo-electron microscopy maps suggests a rationale for the plus-end directional bias associated with the kinesin catalytic core. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1i6i.cif.gz | 88.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1i6i.ent.gz | 65 KB | Display | PDB format |
| PDBx/mmJSON format | 1i6i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/i6/1i6iftp://data.pdbj.org/pub/pdb/validation_reports/i6/1i6i | HTTPS FTP |
|---|
-Related structure data












-Links
 PDBj
PDBj













- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 41102.180 Da / Num. of mol.: 1 / Fragment: MOTOR DOMAIN / Mutation: P202A Source method: isolated from a genetically manipulated source Source: (gene. exp.)  |
|---|---|
| #2: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Mg |
| #3: Chemical | ChemComp-ACP / 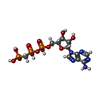  Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM Mass: 505.208 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C11H18N5O12P3 / Comment: AMP-PCP, energy-carrying molecule analogue*YM |
| #4: Chemical | ChemComp-TRS /   Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM Mass: 122.143 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C4H12NO3 / Comment: pH buffer*YM |
| #5: Water | ChemComp-HOH /  Mass: 18.015 Da / Num. of mol.: 231 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 231 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 43.4 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 296 K / Method: vapor diffusion, hanging drop / pH: 6.5 Details: 27% PEG4000, 100MM MES-NAOH PH 6.5, 200MM SODIUM ACETATE. 20MM AMPPCP AND 40MM MGCL2 WERE ADDED 24 HOURS BEFORE COLLECTING X-RAY DATA, VAPOR DIFFUSION, HANGING DROP, temperature 296K | |||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ALS  / Beamline: 5.0.2 / Wavelength: 1.1 Å / Beamline: 5.0.2 / Wavelength: 1.1 Å |
| Detector | Type: ADSC QUANTUM 4 / Detector: CCD / Date: Jul 8, 2000 / Details: mirrors |
| Radiation | Monochromator: double crystal monochromator with Si (111) crystal Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.1 Å / Relative weight: 1 |
| Reflection | Resolution: 1.99→99 Å / Num. all: 25986 / Num. obs: 23596 / % possible obs: 90.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4 % / Biso Wilson estimate: 15.5 Å2 / Rmerge(I) obs: 0.064 / Rsym value: 0.064 / Net I/σ(I): 19.6 |
| Reflection shell | Resolution: 1.99→2.02 Å / Redundancy: 3 % / Rmerge(I) obs: 0.138 / Mean I/σ(I) obs: 8.2 / Num. unique all: 1268 / Rsym value: 0.138 / % possible all: 63 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: catalytic core of therat conventional kinesin Resolution: 2→19.7 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 466354.82 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 / σ(I): 0 / Stereochemistry target values: MLI
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 42.73 Å2 / ksol: 0.358 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→19.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.026 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS σ(F): 0 / % reflection Rfree: 4.9 % / Rfactor obs: 0.219 / Rfactor Rfree: 0.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 32.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.323 / % reflection Rfree: 4.8 % / Rfactor Rwork: 0.271 |
