 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 5u7i | ||||||
|---|---|---|---|---|---|---|---|
| Title | PDE2 catalytic domain complexed with inhibitors | ||||||
 Components Components | cGMP-dependent 3',5'-cyclic phosphodiesterase | ||||||
 Keywords Keywords | Hydrolase/Hydrolase Inhibitor / PDE2 / SBDD / inhibitor / phosphodiesterase / Hydrolase-Hydrolase Inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationcellular response to 2,3,7,8-tetrachlorodibenzodioxine / cellular response to macrophage colony-stimulating factor stimulus / cellular response to cGMP / heart valve development / : / negative regulation of adenylate cyclase-activating G protein-coupled receptor signaling pathway / cellular response to granulocyte macrophage colony-stimulating factor stimulus / positive regulation of vascular permeability / regulation of mitochondrion organization / establishment of endothelial barrier ...cellular response to 2,3,7,8-tetrachlorodibenzodioxine / cellular response to macrophage colony-stimulating factor stimulus / cellular response to cGMP / heart valve development / : / negative regulation of adenylate cyclase-activating G protein-coupled receptor signaling pathway / cellular response to granulocyte macrophage colony-stimulating factor stimulus / positive regulation of vascular permeability / regulation of mitochondrion organization / establishment of endothelial barrier / ventricular septum development / negative regulation of vascular permeability / aorta development / 3',5'-cGMP-stimulated cyclic-nucleotide phosphodiesterase activity / negative regulation of receptor guanylyl cyclase signaling pathway / 3',5'-cyclic-nucleotide phosphodiesterase / cGMP catabolic process / cGMP effects / phosphate ion binding / TPR domain binding / cGMP binding / 3',5'-cyclic-GMP phosphodiesterase activity / monocyte differentiation / 3',5'-cyclic-AMP phosphodiesterase activity / cellular response to transforming growth factor beta stimulus / cAMP binding / negative regulation of cAMP/PKA signal transduction / cellular response to cAMP / synaptic membrane / cellular response to mechanical stimulus / cellular response to xenobiotic stimulus / adenylate cyclase-inhibiting G protein-coupled receptor signaling pathway / positive regulation of inflammatory response / presynaptic membrane / G alpha (s) signalling events / mitochondrial outer membrane / mitochondrial inner membrane / mitochondrial matrix / positive regulation of gene expression / perinuclear region of cytoplasm / Golgi apparatus / magnesium ion binding / negative regulation of transcription by RNA polymerase II / endoplasmic reticulum / protein homodimerization activity / zinc ion binding / identical protein binding / nucleus / plasma membrane / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Pandit, J. | ||||||
 Citation Citation | Journal: J. Med. Chem. / Year: 2017 Title: Application of Structure-Based Design and Parallel Chemistry to Identify a Potent, Selective, and Brain Penetrant Phosphodiesterase 2A Inhibitor. Authors: Helal, C.J. / Arnold, E.P. / Boyden, T.L. / Chang, C. / Chappie, T.A. / Fennell, K.F. / Forman, M.D. / Hajos, M. / Harms, J.F. / Hoffman, W.E. / Humphrey, J.M. / Kang, Z. / Kleiman, R.J. / ...Authors: Helal, C.J. / Arnold, E.P. / Boyden, T.L. / Chang, C. / Chappie, T.A. / Fennell, K.F. / Forman, M.D. / Hajos, M. / Harms, J.F. / Hoffman, W.E. / Humphrey, J.M. / Kang, Z. / Kleiman, R.J. / Kormos, B.L. / Lee, C.W. / Lu, J. / Maklad, N. / McDowell, L. / Mente, S. / O'Connor, R.E. / Pandit, J. / Piotrowski, M. / Schmidt, A.W. / Schmidt, C.J. / Ueno, H. / Verhoest, P.R. / Yang, E.X. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 5u7i.cif.gz | 563.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb5u7i.ent.gz | 463.6 KB | Display | PDB format |
| PDBx/mmJSON format | 5u7i.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/u7/5u7iftp://data.pdbj.org/pub/pdb/validation_reports/u7/5u7i | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  5u7dC  5u7jC  5u7kC  5u7lC  3ituS C: citing same article ( S: Starting model for refinement |
|---|---|
| Similar structure data |










-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| 3 | 
| ||||||||
| 4 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 40225.102 Da / Num. of mol.: 4 / Fragment: Catalytic domain of PDE2, UNP residues 323-663 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Gene: PDE2A / Production host:   Spodoptera frugiperda (fall armyworm) / Strain (production host): Sf21 Spodoptera frugiperda (fall armyworm) / Strain (production host): Sf21References: UniProt: O00408, 3',5'-cyclic-nucleotide phosphodiesterase #2: Chemical | ChemComp-7XS / 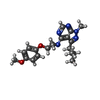  Mass: 367.445 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H25N5O2 Mass: 367.445 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C20H25N5O2#3: Chemical | ChemComp-ZN /   Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Zn#4: Chemical | ChemComp-MG /   Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg Mass: 24.305 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: Mg#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 853 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 853 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.17 Å3/Da / Density % sol: 43.38 % |
|---|---|
| Crystal grow | Temperature: 294 K / Method: vapor diffusion, hanging drop / Details: 25% PEG 3350, 0.1 M Tris, pH 8.5, and 0.2 M MgCl2 |
-Data collection
| Diffraction | Mean temperature: 100 K | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: APS  / Beamline: 17-ID / Wavelength: 1 Å / Beamline: 17-ID / Wavelength: 1 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Detector | Type: ADSC QUANTUM 210 / Detector: CCD / Date: Jun 9, 2006 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection | Resolution: 2→50 Å / Num. obs: 88454 / % possible obs: 97.1 % / Redundancy: 3.6 % / Biso Wilson estimate: 22.73 Å2 / Rmerge(I) obs: 0.053 / Χ2: 1.241 / Net I/σ(I): 15.1 / Num. measured all: 314700 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Reflection shell |
|
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: 3ITU Resolution: 2→34.33 Å / Cor.coef. Fo:Fc: 0.9276 / Cor.coef. Fo:Fc free: 0.9076 / SU R Cruickshank DPI: 0.21 / Cross valid method: THROUGHOUT / σ(F): 0 / SU R Blow DPI: 0.21 / SU Rfree Blow DPI: 0.162 / SU Rfree Cruickshank DPI: 0.163
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso max: 107.55 Å2 / Biso mean: 27.34 Å2 / Biso min: 3.02 Å2
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.259 Å | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: final / Resolution: 2→34.33 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.05 Å / Rfactor Rfree error: 0 / Total num. of bins used: 20
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS params. | Method: refined / Refine-ID: X-RAY DIFFRACTION
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement TLS group |
|
