 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1i6i | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF THE KIF1A MOTOR DOMAIN COMPLEXED WITH MG-AMPPCP | ||||||
 要素 要素 | KINESIN-LIKE PROTEIN KIF1A | ||||||
 キーワード キーワード |  TRANSPORT PROTEIN (運搬体タンパク質) / kinesin (キネシン) / motor protein (モータータンパク質) / catalytic core TRANSPORT PROTEIN (運搬体タンパク質) / kinesin (キネシン) / motor protein (モータータンパク質) / catalytic core | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報protein transport along microtubule / neuronal dense core vesicle membrane / interkinetic nuclear migration / dense core granule cytoskeletal transport / anterograde neuronal dense core vesicle transport / retrograde neuronal dense core vesicle transport / Kinesins / regulation of dendritic spine development / COPI-dependent Golgi-to-ER retrograde traffic / cytoskeleton-dependent intracellular transport ...protein transport along microtubule / neuronal dense core vesicle membrane / interkinetic nuclear migration / dense core granule cytoskeletal transport / anterograde neuronal dense core vesicle transport / retrograde neuronal dense core vesicle transport / Kinesins / regulation of dendritic spine development / COPI-dependent Golgi-to-ER retrograde traffic / cytoskeleton-dependent intracellular transport / regulation of dendritic spine morphogenesis / anterograde axonal transport / plus-end-directed microtubule motor activity / kinesin complex / microtubule-based movement / neuronal dense core vesicle / axon cytoplasm / vesicle-mediated transport / シナプス小胞 / presynapse / microtubule binding / postsynapse / 微小管 / neuron projection / 神経繊維 / 樹状突起 / neuronal cell body / perinuclear region of cytoplasm / ATP hydrolysis activity / protein-containing complex / ATP binding / identical protein binding類似検索 - 分子機能 | ||||||
| 生物種 |  Mus musculus (ハツカネズミ) Mus musculus (ハツカネズミ) | ||||||
| 手法 | X線回折 / シンクロトロン / 分子置換 / 解像度: 2 Å | ||||||
 データ登録者 データ登録者 | Kikkawa, M. / Sablin, E.P. / Okada, Y. / Yajima, H. / Fletterick, R.J. / Hirokawa, N. | ||||||
 引用 引用 | ジャーナル: Nature / 年: 2001 タイトル: Switch-based mechanism of kinesin motors. 著者: M Kikkawa / E P Sablin / Y Okada / H Yajima / R J Fletterick / N Hirokawa /  要旨: Kinesin motors are specialized enzymes that use hydrolysis of ATP to generate force and movement along their cellular tracks, the microtubules. Although numerous biochemical and biophysical studies ...Kinesin motors are specialized enzymes that use hydrolysis of ATP to generate force and movement along their cellular tracks, the microtubules. Although numerous biochemical and biophysical studies have accumulated much data that link microtubule-assisted ATP hydrolysis to kinesin motion, the structural view of kinesin movement remains unclear. This study of the monomeric kinesin motor KIF1A combines X-ray crystallography and cryo-electron microscopy, and allows analysis of force-generating conformational changes at atomic resolution. The motor is revealed in its two functionally critical states-complexed with ADP and with a non-hydrolysable analogue of ATP. The conformational change observed between the ADP-bound and the ATP-like structures of the KIF1A catalytic core is modular, extends to all kinesins and is similar to the conformational change used by myosin motors and G proteins. Docking of the ADP-bound and ATP-like crystallographic models of KIF1A into the corresponding cryo-electron microscopy maps suggests a rationale for the plus-end directional bias associated with the kinesin catalytic core. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1i6i.cif.gz | 88.8 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1i6i.ent.gz | 65 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1i6i.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/i6/1i6iftp://data.pdbj.org/pub/pdb/validation_reports/i6/1i6i | HTTPS FTP |
|---|
-関連構造データ












-リンク
 PDBj
PDBj










- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 41102.180 Da / 分子数: 1 / 断片: MOTOR DOMAIN / 変異: P202A / 由来タイプ: 組換発現 / 由来: (組換発現) Mus musculus (ハツカネズミ) / 遺伝子: KIF1A / プラスミド: PET21B / 生物種 (発現宿主): Escherichia coli / 発現宿主:  Escherichia coli BL21(DE3) (大腸菌) / 株 (発現宿主): BL21(DE3) / 参照: UniProt: P33173 Escherichia coli BL21(DE3) (大腸菌) / 株 (発現宿主): BL21(DE3) / 参照: UniProt: P33173 |
|---|---|
| #2: 化合物 | ChemComp-MG /   分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg 分子量: 24.305 Da / 分子数: 1 / 由来タイプ: 合成 / 式: Mg |
| #3: 化合物 | ChemComp-ACP / 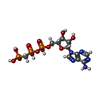  分子量: 505.208 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C11H18N5O12P3 分子量: 505.208 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C11H18N5O12P3コメント: AMP-PCP, エネルギー貯蔵分子類似体*YM |
| #4: 化合物 | ChemComp-TRS / トリスヒドロキシメチルアミノメタン  分子量: 122.143 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H12NO3 / コメント: pH緩衝剤*YM 分子量: 122.143 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C4H12NO3 / コメント: pH緩衝剤*YM |
| #5: 水 | ChemComp-HOH / 水 分子量: 18.015 Da / 分子数: 231 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 231 / 由来タイプ: 天然 / 式: H2O |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 2.3 Å3/Da / 溶媒含有率: 43.4 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 296 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 6.5 詳細: 27% PEG4000, 100MM MES-NAOH PH 6.5, 200MM SODIUM ACETATE. 20MM AMPPCP AND 40MM MGCL2 WERE ADDED 24 HOURS BEFORE COLLECTING X-RAY DATA, VAPOR DIFFUSION, HANGING DROP, temperature 296K | |||||||||||||||||||||||||
| 結晶化 | *PLUS 手法: 蒸気拡散法 | |||||||||||||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: ALS  / ビームライン: 5.0.2 / 波長: 1.1 Å / ビームライン: 5.0.2 / 波長: 1.1 Å |
| 検出器 | タイプ: ADSC QUANTUM 4 / 検出器: CCD / 日付: 2000年7月8日 / 詳細: mirrors |
| 放射 | モノクロメーター: double crystal monochromator with Si (111) crystal プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 1.1 Å / 相対比: 1 |
| 反射 | 解像度: 1.99→99 Å / Num. all: 25986 / Num. obs: 23596 / % possible obs: 90.8 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 4 % / Biso Wilson estimate: 15.5 Å2 / Rmerge(I) obs: 0.064 / Rsym value: 0.064 / Net I/σ(I): 19.6 |
| 反射 シェル | 解像度: 1.99→2.02 Å / 冗長度: 3 % / Rmerge(I) obs: 0.138 / Mean I/σ(I) obs: 8.2 / Num. unique all: 1268 / Rsym value: 0.138 / % possible all: 63 |
- 解析
解析
| ソフトウェア |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: 分子置換 開始モデル: catalytic core of therat conventional kinesin 解像度: 2→19.7 Å / Rfactor Rfree error: 0.007 / Data cutoff high absF: 466354.82 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / 交差検証法: THROUGHOUT / σ(F): 0 / σ(I): 0 / 立体化学のターゲット値: MLI
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: FLAT MODEL / Bsol: 42.73 Å2 / ksol: 0.358 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 32.7 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 2→19.7 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | 解像度: 2→2.13 Å / Rfactor Rfree error: 0.026 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: CNS / バージョン: 1 / 分類: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 精密化 | *PLUS σ(F): 0 / % reflection Rfree: 4.9 % / Rfactor obs: 0.219 / Rfactor Rfree: 0.23 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS Biso mean: 32.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS精密化 シェル | *PLUS Rfactor Rfree: 0.323 / % reflection Rfree: 4.8 % / Rfactor Rwork: 0.271 |
