 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: PDB / ID: 1hvb | ||||||
|---|---|---|---|---|---|---|---|
| タイトル | CRYSTAL STRUCTURE OF STREPTOMYCES R61 DD-PEPTIDASE COMPLEXED WITH A NOVEL CEPHALOSPORIN ANALOG OF CELL WALL PEPTIDOGLYCAN | ||||||
 要素 要素 | D-ALANYL-D-ALANINE CARBOXYPEPTIDASE | ||||||
 キーワード キーワード | HYDROLASE / Protein-cephalosporin complex | ||||||
| 機能・相同性 |  機能・相同性情報 機能・相同性情報serine-type D-Ala-D-Ala carboxypeptidase / serine-type D-Ala-D-Ala carboxypeptidase activity / peptidoglycan biosynthetic process / cell wall organization / regulation of cell shape / proteolysis / extracellular region 類似検索 - 分子機能 | ||||||
| 生物種 |  Streptomyces sp. (バクテリア) Streptomyces sp. (バクテリア) | ||||||
| 手法 |  X線回折 / シンクロトロン / フーリエ合成 / 解像度: 1.17 Å X線回折 / シンクロトロン / フーリエ合成 / 解像度: 1.17 Å | ||||||
 データ登録者 データ登録者 | McDonough, M.A. / Lee, W. / Silvaggi, N.R. / Mobashery, S. / Kelly, J.A. | ||||||
 引用 引用 | ジャーナル: Proc.Natl.Acad.Sci.USA / 年: 2001 タイトル: A 1.2-A snapshot of the final step of bacterial cell wall biosynthesis. 著者: Lee, W. / McDonough, M.A. / Kotra, L. / Li, Z.H. / Silvaggi, N.R. / Takeda, Y. / Kelly, J.A. / Mobashery, S. #1: ジャーナル: Biochemistry / 年: 1995タイトル: Binding of Cephalothin and Cefotaxime to D-Ala-D-Ala-Peptidase Reveals A Functional Basis of a Natural Mutation in a Low-Affinity Penicillin-Binding Protein and in Extended-Spectrum Beta-Lactamases 著者: Kuzin, A.P. / Liu, H. / Kelly, J.A. / Knox, J.R. #2: ジャーナル: J.Mol.Biol. / 年: 1995タイトル: The Refined Crystallographic Structure of a DD-Peptidase Penicillin-target Enzyme at 1.6 Angstrom Resolution 著者: Kelly, J.A. / Kuzin, A.P. | ||||||
| 履歴 |
|
- 構造の表示
構造の表示
| 構造ビューア | 分子: MolmilJmol/JSmol |
|---|
- ダウンロードとリンク
ダウンロードとリンク
-ダウンロード
| PDBx/mmCIF形式 | 1hvb.cif.gz | 179.2 KB | 表示 | PDBx/mmCIF形式 |
|---|---|---|---|---|
| PDB形式 | pdb1hvb.ent.gz | 141.4 KB | 表示 | PDB形式 |
| PDBx/mmJSON形式 | 1hvb.json.gz | ツリー表示 | PDBx/mmJSON形式 | |
| その他 |  その他のダウンロード その他のダウンロード |
-検証レポート
| アーカイブディレクトリ | https://data.pdbj.org/pub/pdb/validation_reports/hv/1hvbftp://data.pdbj.org/pub/pdb/validation_reports/hv/1hvb | HTTPS FTP |
|---|
-関連構造データ
| 関連構造データ |  3pteS S: 精密化の開始モデル |
|---|---|
| 類似構造データ |












-リンク
 PDBj
PDBj
- 集合体
集合体
| 登録構造単位 | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| 単位格子 |
|
-要素
| #1: タンパク質 | 分子量: 37422.574 Da / 分子数: 1 / 由来タイプ: 天然 / 由来: (天然) Streptomyces sp. (バクテリア) / 株: R61参照: UniProt: P15555, serine-type D-Ala-D-Ala carboxypeptidase |
|---|---|
| #2: 化合物 | ChemComp-CEH / 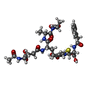  分子量: 750.773 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C32H42N6O13S 分子量: 750.773 Da / 分子数: 1 / 由来タイプ: 合成 / 式: C32H42N6O13S |
| #3: 水 | ChemComp-HOH /  分子量: 18.015 Da / 分子数: 555 / 由来タイプ: 天然 / 式: H2O 分子量: 18.015 Da / 分子数: 555 / 由来タイプ: 天然 / 式: H2O |
| Has protein modification | Y |
-実験情報
-実験
| 実験 | 手法: X線回折 / 使用した結晶の数: 1 |
|---|
- 試料調製
試料調製
| 結晶 | マシュー密度: 1.94 Å3/Da / 溶媒含有率: 36.6 % | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 結晶化 | 温度: 298 K / 手法: 蒸気拡散法, ハンギングドロップ法 / pH: 6.8 詳細: PEG 8000, sodium phosphate, pH 6.8, VAPOR DIFFUSION, HANGING DROP, temperature 298K | |||||||||||||||
| 結晶化 | *PLUS 手法: unknown / 詳細: Kuzin, A.P., (1995) Biochemistry, 34, 9532. | |||||||||||||||
| 溶液の組成 | *PLUS
|
-データ収集
| 回折 | 平均測定温度: 100 K |
|---|---|
| 放射光源 | 由来: シンクロトロン / サイト: NSLS  / ビームライン: X12C / 波長: 0.979 Å / ビームライン: X12C / 波長: 0.979 Å |
| 検出器 | タイプ: BRANDEIS - B4 / 検出器: CCD / 日付: 2000年1月29日 / 詳細: Monochromator, Mirror |
| 放射 | モノクロメーター: Si(111) / プロトコル: SINGLE WAVELENGTH / 単色(M)・ラウエ(L): M / 散乱光タイプ: x-ray |
| 放射波長 | 波長: 0.979 Å / 相対比: 1 |
| 反射 | 解像度: 1.17→20 Å / Num. all: 2330719 / Num. obs: 111370 / % possible obs: 95.9 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / 冗長度: 5 % / Biso Wilson estimate: 18.6 Å2 / Rmerge(I) obs: 0.04 / Net I/σ(I): 25 |
| 反射 シェル | 解像度: 1.17→1.21 Å / Rmerge(I) obs: 0.162 / Mean I/σ(I) obs: 4.6 / Num. unique all: 8392 / % possible all: 72.9 |
| 反射 | *PLUS Num. measured all: 2330719 / Rmerge(I) obs: 0.04 |
| 反射 シェル | *PLUS % possible obs: 73 % |
- 解析
解析
| ソフトウェア |
| |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 精密化 | 構造決定の手法: フーリエ合成 開始モデル: PDB ENTRY 3PTE 解像度: 1.17→10 Å / Num. parameters: 29978 / Num. restraintsaints: 36742 / Isotropic thermal model: ISOTROPIC / 交差検証法: free r / σ(F): 0 / 立体化学のターゲット値: ENGH AND HUBER 詳細: THE ESTIMATED STANDARD DEVIATION OF ATOMIC COORDINATES FOR ALL ATOMS IS 0.032 ANGSTROMS DETERMINED BY INVERSION OF THE LEAST-SQUARES MATRIX. ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) ...詳細: THE ESTIMATED STANDARD DEVIATION OF ATOMIC COORDINATES FOR ALL ATOMS IS 0.032 ANGSTROMS DETERMINED BY INVERSION OF THE LEAST-SQUARES MATRIX. ANISOTROPIC REFINEMENT REDUCED FREE R (NO CUTOFF) BY 3.5%. WATER MOLECULES DESIGNATED AS B CONFORMATION ARE PRESENT IN UN-LIGAND BOUND MOLECULES IN THE CRYSTAL.
| |||||||||||||||||||||||||||||||||
| 溶媒の処理 | 溶媒モデル: MOEWS & KRETSINGER, J.MOL.BIOL.91(1973)201-228 | |||||||||||||||||||||||||||||||||
| 原子変位パラメータ | Biso mean: 16.1 Å2 | |||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.146 Å / Luzzati d res low obs: 20 Å / Num. disordered residues: 35 / Occupancy sum hydrogen: 268.92 / Occupancy sum non hydrogen: 3181.8 | |||||||||||||||||||||||||||||||||
| 精密化ステップ | サイクル: LAST / 解像度: 1.17→10 Å
| |||||||||||||||||||||||||||||||||
| 拘束条件 |
| |||||||||||||||||||||||||||||||||
| ソフトウェア | *PLUS 名称: SHELXL-97 / 分類: refinement | |||||||||||||||||||||||||||||||||
| 精密化 | *PLUS σ(F): 0 / % reflection Rfree: 5 % | |||||||||||||||||||||||||||||||||
| 溶媒の処理 | *PLUS | |||||||||||||||||||||||||||||||||
| 原子変位パラメータ | *PLUS | |||||||||||||||||||||||||||||||||
| 拘束条件 | *PLUS
|
