 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h9r | ||||||
|---|---|---|---|---|---|---|---|
| Title | Tungstate bound complex Dimop domain of ModE from E.coli | ||||||
 Components Components | MOLYBDENUM TRANSPORT PROTEIN MODE | ||||||
 Keywords Keywords | TRANSCRIPTION REGULATOR | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of DNA-templated transcription initiation / molybdate ion transport / molybdenum ion binding / cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / DNA-templated transcription / DNA binding ...negative regulation of DNA-templated transcription initiation / molybdate ion transport / molybdenum ion binding / cis-regulatory region sequence-specific DNA binding / DNA-binding transcription factor activity / negative regulation of DNA-templated transcription / regulation of DNA-templated transcription / positive regulation of DNA-templated transcription / DNA-templated transcription / DNA binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 1.9 Å | ||||||
 Authors Authors | Gourley, D.G. / Hunter, W.N. | ||||||
 Citation Citation | Journal: J.Biol.Chem. / Year: 2001 Title: Oxyanion Binding Alters Conformational and Quaternary Structure of the C-Terminal Domain of the Transcriptional Regulator Mode; Implications for Molybdate-Dependant Regulation, Signalling, Storage and Transport Authors: Gourley, D.G. / Schuttelkopf, A.W. / Anderson, L.A. / Price, N.C. / Boxer, D.H. / Hunter, W.N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h9r.cif.gz | 74 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h9r.ent.gz | 53.7 KB | Display | PDB format |
| PDBx/mmJSON format | 1h9r.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h9/1h9rftp://data.pdbj.org/pub/pdb/validation_reports/h9/1h9r | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1h9sC  1b9nS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |











-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 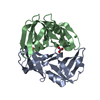
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.125, -0.974, 0.187), Vector: |
-Components
| #1: Protein | Mass: 15048.901 Da / Num. of mol.: 2 / Fragment: MOLYBDATE BINDING DOMAIN RESIDUE 123-262 / Mutation: YES Source method: isolated from a genetically manipulated source Source: (gene. exp.) #2: Chemical |   Mass: 247.838 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: WO4 Mass: 247.838 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: WO4#3: Chemical | ChemComp-NI / |   Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni Mass: 58.693 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ni#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 312 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 312 / Source method: isolated from a natural source / Formula: H2OCompound details | CHAIN A, B ENGINEERED MUTATION FROM LEU 123 MET BINDS MOLYBDENUM. REGULATES MOD OPERON BY BINDING ...CHAIN A, B ENGINEERED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.19 Å3/Da / Density % sol: 43.77 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.6 / Details: pH 7.60 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IV / Detector: IMAGE PLATE / Details: MIRRORS |
| Radiation | Monochromator: NI FILTER / Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→30 Å / Num. obs: 20410 / % possible obs: 97.9 % / Redundancy: 9 % / Biso Wilson estimate: 22 Å2 / Rsym value: 0.49 / Net I/σ(I): 27.8 |
| Reflection | *PLUS Highest resolution: 1.9 Å / Lowest resolution: 30 Å / Num. measured all: 182781 / Rmerge(I) obs: 0.049 |
| Reflection shell | *PLUS % possible obs: 78.9 % / Rmerge(I) obs: 0.147 / Mean I/σ(I) obs: 9.5 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1B9N Resolution: 1.9→20 Å / SU B: 3.285 / SU ML: 0.1 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.166 / ESU R Free: 0.155
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.2 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.17 / Rfactor Rwork: 0.17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS |
