 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h66: CRYSTAL STRUCTURE OF HUMAN NAD[P]H-QUINONE OXIDOREDUCTASE CO WITH... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h66 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN NAD[P]H-QUINONE OXIDOREDUCTASE CO WITH 2,5-diaziridinyl-3-hydroxyl-6-methyl-1,4-benzoquinone | ||||||
 Components Components | NAD(P)H DEHYDROGENASE [QUINONE] 1 | ||||||
 Keywords Keywords | OXIDOREDUCTASE / FLAVOPROTEIN / ROSSMANN FOLD | ||||||
| Function / homology |  Function and homology information Function and homology informationubiquinone metabolic process / vitamin E metabolic process / NAD(P)H dehydrogenase (quinone) / NADPH dehydrogenase (quinone) activity / cytochrome-b5 reductase activity, acting on NAD(P)H / vitamin K metabolic process / NADH dehydrogenase (quinone) (non-electrogenic) activity / NAD(P)H dehydrogenase (quinone) activity / NFE2L2 regulating anti-oxidant/detoxification enzymes / Regulation of ornithine decarboxylase (ODC) ...ubiquinone metabolic process / vitamin E metabolic process / NAD(P)H dehydrogenase (quinone) / NADPH dehydrogenase (quinone) activity / cytochrome-b5 reductase activity, acting on NAD(P)H / vitamin K metabolic process / NADH dehydrogenase (quinone) (non-electrogenic) activity / NAD(P)H dehydrogenase (quinone) activity / NFE2L2 regulating anti-oxidant/detoxification enzymes / Regulation of ornithine decarboxylase (ODC) / synaptic transmission, cholinergic / negative regulation of ferroptosis / nitric oxide biosynthetic process / removal of superoxide radicals / xenobiotic metabolic process / cell redox homeostasis / protein catabolic process / negative regulation of protein catabolic process / response to toxic substance / protein polyubiquitination / response to lipopolysaccharide / cellular response to oxidative stress / response to oxidative stress / innate immune response / synapse / RNA binding / identical protein binding / nucleus / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  HOMO SAPIENS (human) HOMO SAPIENS (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Faig, M. / Bianchet, M.A. / Winski, S. / Ross, D. / Amzel, L.M. | ||||||
 Citation Citation | Journal: Structure / Year: 2001 Title: Structure-Based Development of Anticancer Drugs: Complexes of Nad(P)H:Quinone Oxidoreductase 1 with Chemotherapeutic Quinones Authors: Faig, M. / Bianchet, M.A. / Winski, S. / Hargreaves, R. / Moody, C.J. / Hudnott, A.R. / Ross, D. / Amzel, L.M. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 2000Title: Structures of Recombinant Mouse and Human Nad(P)H:Quinone Oxidoreductases:Species Comparison and Structural Changes with Substrate Binding and Release Authors: Faig, M. / Bianchet, M.A. / Chen, S. / Winski, S. / Ross, D. / Talalay, P. / Amzel, L.M. #2: Journal: Biochem.Soc.Trans. / Year: 1999 Title: Structure and Mechanism of Cytosolic Quinone Reductase Authors: Bianchet, M.A. / Foster, C. / Faig, M. / Talalay, P. / Amzel, L.M. #3: Journal: Biochemistry / Year: 1999Title: Crystal Structure of Human Quinone Reductase Type 2, a Metalloprotein Authors: Foster, C. / Bianchet, M.A. / Talalay, P. / Zhao, Q. / Amzel, L.M. #4: Journal: Proc.Natl.Acad.Sci.USA / Year: 1995Title: The Three-Dimensional Structure of Nad(P)H:Quinone Reductase, a Flavoprotein Involved in Cancer Chemoprotection and Chemotherapy: Mechanism of Two-Electron Reduction Authors: Li, R. / Bianchet, M.A. / Talalay, P. / Amzel, L.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h66.cif.gz | 241.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h66.ent.gz | 196.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1h66.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h6/1h66ftp://data.pdbj.org/pub/pdb/validation_reports/h6/1h66 | HTTPS FTP |
|---|
-Related structure data















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 30748.361 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) HOMO SAPIENS (human) / Production host:  #2: Chemical | ChemComp-RH1 / 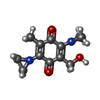  Mass: 234.251 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C12H14N2O3 Mass: 234.251 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C12H14N2O3#3: Chemical | ChemComp-FAD /   Mass: 785.550 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM Mass: 785.550 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C27H33N9O15P2 / Comment: FAD*YM#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 701 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 701 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.41 Å3/Da / Density % sol: 48.97 % | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 8.5 / Details: pH 8.50 | ||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 25 ℃ / pH: 8 / Method: vapor diffusion, hanging drop / Details: Faig, M., (2000) Proc.Natl.Acad.Sci.USA, 97, 3177. | ||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: NSLS  / Beamline: X25 / Wavelength: 1 / Beamline: X25 / Wavelength: 1 |
| Detector | Type: BRANDEIS CCD / Detector: CCD |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 |
| Reflection | Resolution: 2→19.96 Å / Num. obs: 73718 / % possible obs: 93.8 % / Observed criterion σ(I): 0 / Redundancy: 2.5 % / Biso Wilson estimate: 9.7 Å2 / Rmerge(I) obs: 0.09 |
| Reflection | *PLUS Redundancy: 3.9 % / Rmerge(I) obs: 0.091 |
| Reflection shell | *PLUS % possible obs: 83.8 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT / Resolution: 2→19.96 Å / Rfactor Rfree error: 0.001 / Data cutoff high absF: 478391.8 / Data cutoff low absF: 0 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 34.4807 Å2 / ksol: 0.343615 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 22.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→19.96 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.003 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: CNS / Version: 1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.199 / Rfactor Rfree: 0.256 / Rfactor Rwork: 0.199 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
