 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1h3f: Tyrosyl-tRNA synthetase from Thermus thermophilus complexed with ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h3f | ||||||
|---|---|---|---|---|---|---|---|
| Title | Tyrosyl-tRNA synthetase from Thermus thermophilus complexed with tyrosinol | ||||||
 Components Components | TYROSYL-TRNA SYNTHETASE | ||||||
 Keywords Keywords | LIGASE / AMINOACYL-TRNA SYNTHETASE / ATP + L-TYROSINE + TRNA(TYR)->AMP + PPI + L-TYROSYL-TRNA(TY CLASS I AMINOACYL-TRNA SYNTHETASE | ||||||
| Function / homology |  Function and homology information Function and homology informationtyrosyl-tRNA aminoacylation / tyrosine-tRNA ligase / tyrosine-tRNA ligase activity / RNA binding / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |   THERMUS THERMOPHILUS (bacteria) THERMUS THERMOPHILUS (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 2 Å | ||||||
 Authors Authors | Cusack, S. / Yaremchuk, A. / Kriklivyi, I. / Tukalo, M. | ||||||
 Citation Citation | Journal: Embo J. / Year: 2002 Title: Class I Tyrosyl-tRNA Synthetase Has a Class II Mode or tRNA Recognition Authors: Yaremchuk, A. / Kriklivyi, I. / Tukalo, M. / Cusack, S. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h3f.cif.gz | 175.7 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h3f.ent.gz | 140.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1h3f.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h3/1h3fftp://data.pdbj.org/pub/pdb/validation_reports/h3/1h3f | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||
| Unit cell |
| ||||||||||||
| Noncrystallographic symmetry (NCS) | NCS oper:
|
-Components
| #1: Protein | Mass: 48786.203 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) THERMUS THERMOPHILUS (bacteria) / Strain: HB27 / Production host: #2: Chemical | ChemComp-SO4 /   Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 5 / Source method: obtained synthetically / Formula: SO4#3: Chemical | 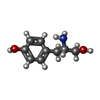  Type: L-peptide linking / Mass: 167.205 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H13NO2 Type: L-peptide linking / Mass: 167.205 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C9H13NO2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 254 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 254 / Source method: isolated from a natural source / Formula: H2OCompound details | CATALYTIC ACTIVITY: ATP + L-TYROSINE + TRNA(TYR) = AMP + DIPHOSPHATE + L-TYROSYL-TRNA(TYR). ...CATALYTIC ACTIVITY: ATP + L-TYROSINE + TRNA(TYR) = AMP + DIPHOSPHAT | Sequence details | UNMODELLED | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 54.6 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.8 Details: 12MG/ML PROTEIN IN 1:1 RATIO WITH RESERVOIR SOLUTION CONTAINING 1.2M AMMONIUM SULPHATE, 50MM MES(PH5.8), 10MM MGCL2, 0.5MM DDT, pH 5.80 | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293 K / Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-1 / Wavelength: 0.934 / Beamline: ID14-1 / Wavelength: 0.934 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Oct 15, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.934 Å / Relative weight: 1 |
| Reflection | Resolution: 2→25 Å / Num. obs: 70951 / % possible obs: 97.6 % / Redundancy: 6.5 % / Biso Wilson estimate: 17.3 Å2 / Rmerge(I) obs: 0.089 / Net I/σ(I): 5.7 |
| Reflection shell | Resolution: 2→2.05 Å / Redundancy: 2.4 % / Rmerge(I) obs: 0.306 / Mean I/σ(I) obs: 2.3 / % possible all: 84.5 |
| Reflection | *PLUS Lowest resolution: 25 Å / Num. measured all: 464280 |
| Reflection shell | *PLUS % possible obs: 84.5 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: T. THERMOPHILUS TYROSYL-TRNA SYNTHETASE PREVIOUSLY DETERMINED BY SIRAS BY SAME AUTHORS Resolution: 2→25 Å / Rfactor Rfree error: 0.004 / Data cutoff high absF: 1834546.75 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 0 Details: CHAIN A RESIDUES 1-4, 80-100 AND 348-351 AND CHAIN B RESIDUES 1-4, 84-96 AND 345-352 WERE UNMODELED DUE TO DISORDER
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Solvent model: FLAT MODEL / Bsol: 48.6936 Å2 / ksol: 0.38785 e/Å3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 32.5 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→25 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.13 Å / Rfactor Rfree error: 0.013 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Lowest resolution: 20 Å / Num. reflection Rfree: 3557 / % reflection Rfree: 4.9 % / Rfactor Rfree: 0.26 / Rfactor Rwork: 0.233 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rwork: 0.28 |
