 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1h3d | ||||||
|---|---|---|---|---|---|---|---|
| Title | STRUCTURE OF THE E.COLI ATP-PHOSPHORIBOSYLTRANSFERASE | ||||||
 Components Components | ATP-PHOSPHORIBOSYLTRANSFERASE | ||||||
 Keywords Keywords | TRANSFERASE / PHOSPHORIBOSYLTRANSFERASE / HISITIDINE BIOSYNTHESIS / GLYCOSYLTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationATP phosphoribosyltransferase / ATP phosphoribosyltransferase activity / L-histidine biosynthetic process / magnesium ion binding / ATP binding / cytosol / cytoplasm Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.7 Å | ||||||
 Authors Authors | Lohkamp, B. / McDermott, G. / Coggins, J.R. / Lapthorn, A.J. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2004 Title: The Structure of Escherichia Coli ATP-Phosphoribosyltransferase: Identification of Substrate Binding Sites and Mode of AMP Inhibition Authors: Lohkamp, B. / Mcdermott, G. / Campbell, S. / Coggins, J.R. / Lapthorn, A.J. | ||||||
| History |
| ||||||
| Remark 285 | TEXT TO EXPLAIN UNUSUAL UNIT-CELL DATA: HEXAGONAL SETTING USED |
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1h3d.cif.gz | 67.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1h3d.ent.gz | 50.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1h3d.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/h3/1h3dftp://data.pdbj.org/pub/pdb/validation_reports/h3/1h3d | HTTPS FTP |
|---|
-Related structure data







-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | x 6
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 33408.746 Da / Num. of mol.: 1 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P10366, UniProt: P60757*PLUS, ATP phosphoribosyltransferase | ||
|---|---|---|---|
| #2: Chemical | ChemComp-AMP /   Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM Mass: 347.221 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H14N5O7P / Comment: AMP*YM | ||
| #3: Chemical | 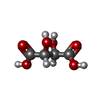  Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6 Mass: 150.087 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C4H6O6Compound details | CATALYTIC ACTIVITY: 1-(5-PHOSPHO-D-RIBOSYL)-ATP + DIPHOSPHATE = ATP + 5-PHOSPHO-ALPHA-D-RIBOSE 1- ...CATALYTIC ACTIVITY: 1-(5-PHOSPHO-D-RIBOSYL)-ATP + DIPHOSPHAT | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.95 Å3/Da / Density % sol: 58 % | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 5.6 Details: PROTEIN WAS CRYSTALLIZED FROM 1.3M SODIUM TARTRATE, 50-200MM MGCL2, 100MM CITRATE BUFFER, PH 5.6, IN PRESENCE OF 2MM AMP | ||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 293 K / pH: 5.6 / Method: vapor diffusion, sitting drop / Details: Lohkamp, B., (2000) Acta Crystallogr., D56, 1488. | ||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX9.6 / Wavelength: 0.87 / Beamline: PX9.6 / Wavelength: 0.87 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Nov 15, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.87 Å / Relative weight: 1 |
| Reflection | Resolution: 2.7→81.65 Å / Num. obs: 10860 / % possible obs: 99.5 % / Observed criterion σ(I): 2 / Redundancy: 6.7 % / Biso Wilson estimate: 83.842 Å2 / Rmerge(I) obs: 0.059 / Net I/σ(I): 18 |
| Reflection shell | Resolution: 2.7→2.75 Å / Redundancy: 6.7 % / Rmerge(I) obs: 0.796 / Mean I/σ(I) obs: 3.1 / % possible all: 100 |
| Reflection | *PLUS Highest resolution: 2.7 Å / Redundancy: 6.7 % / Rmerge(I) obs: 0.059 |
| Reflection shell | *PLUS % possible obs: 100 % / Redundancy: 6.7 % / Rmerge(I) obs: 0.796 / Mean I/σ(I) obs: 3.1 |
- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.7→81.65 Å / SU B: 13.292 / SU ML: 0.2812 / Cross valid method: THROUGHOUT / σ(F): 0 / ESU R: 0.89567 / ESU R Free: 0.36452
| ||||||||||||||||||||
| Displacement parameters | Biso mean: 46.118 Å2
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.7→81.65 Å
| ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2.7 Å / Rfactor Rfree: 0.277 / Rfactor Rwork: 0.222 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||
| Refine LS restraints | *PLUS
|
