 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1gz5 | ||||||
|---|---|---|---|---|---|---|---|
| Title | Trehalose-6-phosphate synthase. OtsA | ||||||
 Components Components | ALPHA-TREHALOSE-PHOSPHATE SYNTHASE | ||||||
 Keywords Keywords | SYNTHASE / TREHALOSE / GLYCOSYLTRANSFERASE / ROSSMANN-FOLD / GLUCOSE-6-PHOSPHATE / TREHALOSE-6-PHOSPHATE | ||||||
| Function / homology |  Function and homology information Function and homology informationalpha,alpha-trehalose-phosphate synthase (UDP-forming) / alpha,alpha-trehalose-phosphate synthase (UDP-forming) activity / trehalose biosynthetic process / response to osmotic stress / cellular response to cold / response to stress / DNA damage response Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.43 Å X-RAY DIFFRACTION / SYNCHROTRON / MAD / Resolution: 2.43 Å | ||||||
 Authors Authors | Gibson, R.P. / Turkenburg, J.P. / Davies, G.J. | ||||||
 Citation Citation | Journal: Chem.Biol. / Year: 2002 Title: Insights Into Trehalose Synthesis Provided by the Structure of the Retaining Glycosyltransferase Otsa Authors: Gibson, R.P. / Turkenburg, J.P. / Charnock, S.J. / Lloyd, R. / Davies, G.J. #1: Journal: Acta Crystallogr.,Sect.D / Year: 2002 Title: Characterization of Escherichia Coli Otsa, a Trehalose-6-Phosphate Synthase from Glycosyltransferase Family 20 Authors: Gibson, R.P. / Lloyd, R.M. / Charnock, S.J. / Davies, G.J. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1gz5.cif.gz | 367.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1gz5.ent.gz | 300.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1gz5.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/gz/1gz5ftp://data.pdbj.org/pub/pdb/validation_reports/gz/1gz5 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|








-Links
 PDBj
PDBj







- Assembly
Assembly
| Deposited unit | 
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Unit cell |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Noncrystallographic symmetry (NCS) | NCS domain:
NCS domain segments: Component-ID: 1 / Refine code: 2
NCS ensembles :
NCS oper:
|
-Components
| #1: Protein | Mass: 51871.273 Da / Num. of mol.: 4 Source method: isolated from a genetically manipulated source Source: (gene. exp.) References: UniProt: P31677, alpha,alpha-trehalose-phosphate synthase (UDP-forming) #2: Chemical | ChemComp-UDP / 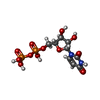  Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM Type: RNA linking / Mass: 404.161 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C9H14N2O12P2 / Comment: UDP*YM#3: Sugar | ChemComp-G6P / 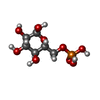  Type: D-saccharide, alpha linking / Mass: 260.136 Da / Num. of mol.: 4 Type: D-saccharide, alpha linking / Mass: 260.136 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H13O9P #4: Chemical | ChemComp-IMD /   Mass: 69.085 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H5N2 Mass: 69.085 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: C3H5N2#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 288 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 288 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.7 Å3/Da / Density % sol: 53.3 % Description: THESE STATISTICS REFLECT SEPARATE TREATMENT OF FREIDEL MATES DURING SCALING PROCEDURE. TRUE MULTIPLICITY IS THUS APPROXIMATALY TWICE THE VALUE GIVEN | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7.5 Details: 14% PEG4000, 0.15M NAAC, 0.1M IMIDAZOLE PH 7.5, 10MM MG-UDP, 10MM G6P | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: ESRF  / Beamline: ID14-4 / Wavelength: 0.9392 / Beamline: ID14-4 / Wavelength: 0.9392 |
| Detector | Type: ADSC CCD / Detector: CCD / Date: Nov 15, 2001 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.9392 Å / Relative weight: 1 |
| Reflection | Resolution: 2.45→20 Å / Num. obs: 86205 / % possible obs: 99.9 % / Redundancy: 2.8 % / Rmerge(I) obs: 0.056 / Net I/σ(I): 17 |
| Reflection shell | Resolution: 2.45→2.54 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.436 / Mean I/σ(I) obs: 2.4 / % possible all: 99.5 |
| Reflection | *PLUS Lowest resolution: 20 Å |
| Reflection shell | *PLUS % possible obs: 99.5 % / Rmerge(I) obs: 0.43 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MAD / Resolution: 2.43→100 Å / Cor.coef. Fo:Fc: 0.953 / Cor.coef. Fo:Fc free: 0.943 / SU B: 9.449 / SU ML: 0.21 / Cross valid method: THROUGHOUT / ESU R: 0.427 / ESU R Free: 0.242 Stereochemistry target values: MAXIMUM LIKELIHOOD WITH PHASES Details: TLS REFINEMENT USED THROUGHOUT. MOLECULE C SHOWS LARGE DISORDER IN ITS N-TERMINAL DOMAIN REFLECTED IN LARGE TLS VALUES AND POOR ELECTRON DENSITY. RESIDUES ALA 208 B AND ALA 208 D COULD NOT ...Details: TLS REFINEMENT USED THROUGHOUT. MOLECULE C SHOWS LARGE DISORDER IN ITS N-TERMINAL DOMAIN REFLECTED IN LARGE TLS VALUES AND POOR ELECTRON DENSITY. RESIDUES ALA 208 B AND ALA 208 D COULD NOT BE BUILT INTO THE ELECTRON DENSITY DUE TO LARGE DISORDER FOUND IN THESE LOOP REGIONS.
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | Ion probe radii: 0.8 Å / Shrinkage radii: 0.8 Å / VDW probe radii: 1.4 Å / Solvent model: BABINET MODEL WITH MASK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 34.54 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.43→100 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
|