 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1fh0: CRYSTAL STRUCTURE OF HUMAN CATHEPSIN V COMPLEXED WITH AN IRREVERS... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1fh0 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF HUMAN CATHEPSIN V COMPLEXED WITH AN IRREVERSIBLE VINYL SULFONE INHIBITOR | ||||||
 Components Components | CATHEPSIN V | ||||||
 Keywords Keywords | HYDROLASE/HYDROLASE inhibitor / cathepsin / papain / protease / cancer / HYDROLASE-HYDROLASE inhibitor complex | ||||||
| Function / homology |  Function and homology information Function and homology informationcathepsin V / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / Trafficking and processing of endosomal TLR / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / extracellular matrix disassembly / Degradation of the extracellular matrix / cysteine-type peptidase activity / MHC class II antigen presentation / lysosomal lumen ...cathepsin V / RUNX1 regulates transcription of genes involved in differentiation of keratinocytes / Trafficking and processing of endosomal TLR / Assembly of collagen fibrils and other multimeric structures / Activation of Matrix Metalloproteinases / extracellular matrix disassembly / Degradation of the extracellular matrix / cysteine-type peptidase activity / MHC class II antigen presentation / lysosomal lumen / : / Endosomal/Vacuolar pathway / antigen processing and presentation of exogenous peptide antigen via MHC class II / lysosome / serine-type endopeptidase activity / cysteine-type endopeptidase activity / : / extracellular region Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.6 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.6 Å | ||||||
 Authors Authors | Somoza, J.R. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 2000 Title: Crystal structure of human cathepsin V. Authors: Somoza, J.R. / Zhan, H. / Bowman, K.K. / Yu, L. / Mortara, K.D. / Palmer, J.T. / Clark, J.M. / McGrath, M.E. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1fh0.cif.gz | 105.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1fh0.ent.gz | 80.3 KB | Display | PDB format |
| PDBx/mmJSON format | 1fh0.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/fh/1fh0ftp://data.pdbj.org/pub/pdb/validation_reports/fh/1fh0 | HTTPS FTP |
|---|
-Related structure data
| Similar structure data |
|---|










-Links
 PDBj
PDBj












- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Details | This enzyme is most likely active as a monomer. |
-Components
| #1: Protein | Mass: 24040.959 Da / Num. of mol.: 2 / Mutation: N108Q, N178D Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Production host:  References: UniProt: O60911, Hydrolases; Acting on peptide bonds (peptidases); Cysteine endopeptidases #2: Chemical | 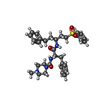  Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 576.749 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C32H40N4O4S Type: peptide-like, Peptide-like / Class: Inhibitor / Mass: 576.749 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C32H40N4O4SDetails: This inhibitor is covalently linked to BNS, 4-SULFONYLBENZENE GROUP, to form an irreversible vinyl sulfone inhibitor. References: MePip-Phe-HphVSPh #3: Chemical | ChemComp-SO4 / |   Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4 Mass: 96.063 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: SO4#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 287 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 287 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | Nonpolymer details | INHIBITOR 4-METHYLPIPERAZINE-1-CARBOXYLIC ACID [1-[(3-BENZENESULFONYL-1-PHENETHYLALLYL)CARBAMOYL]-2- ...INHIBITOR 4-METHYLPIPE | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.9 Å3/Da / Density % sol: 57.54 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 290 K / Method: vapor diffusion, hanging drop / pH: 8.5 Details: Lithium sulfate, PEG 4000, tris, glycerol, pH 8.5, VAPOR DIFFUSION, HANGING DROP, temperature 290.0K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 17 ℃ / pH: 4.5 | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SSRL  / Beamline: BL9-1 / Wavelength: 0.97 / Beamline: BL9-1 / Wavelength: 0.97 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Jun 22, 2000 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 0.97 Å / Relative weight: 1 |
| Reflection | Resolution: 1.6→100 Å / Num. all: 78054 / Num. obs: 78054 / % possible obs: 98.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 5 % / Biso Wilson estimate: 20 Å2 / Rmerge(I) obs: 0.038 / Net I/σ(I): 37.3 |
| Reflection shell | Resolution: 1.6→1.63 Å / Redundancy: 5 % / Rmerge(I) obs: 0.424 / Num. unique all: 3539 / % possible all: 89.9 |
| Reflection | *PLUS Highest resolution: 1.6 Å / Num. measured all: 386837 |
| Reflection shell | *PLUS Highest resolution: 1.6 Å / % possible obs: 89.9 % / Mean I/σ(I) obs: 3.45 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.6→50 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh and Huber
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.6→50 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: CNX / Classification: refinement | |||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.6 Å / Lowest resolution: 50 Å / σ(F): 0 / Rfactor obs: 0.197 | |||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
