 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1f50: BACTERIORHODOPSIN-BR STATE OF THE E204Q MUTANT AT 1.7 ANGSTROM RE... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1f50 | ||||||
|---|---|---|---|---|---|---|---|
| Title | BACTERIORHODOPSIN-BR STATE OF THE E204Q MUTANT AT 1.7 ANGSTROM RESOLUTION | ||||||
 Components Components | BACTERIORHODOPSIN | ||||||
 Keywords Keywords | PROTON TRANSPORT / MEMBRANE PROTEIN / ION PUMP / RETINAL PROTEIN / LIPIDS / PHOTORECEPTOR / HALOARCHAEA / 7-TRANSMEMBRANE / ION TRANSPORT / MEROHEDRAL TWINNING / E204Q mutant ground state / photocycle intermediate | ||||||
| Function / homology |  Function and homology information Function and homology informationlight-driven active monoatomic ion transmembrane transporter activity / photoreceptor activity / phototransduction / monoatomic ion channel activity / proton transmembrane transport / plasma membrane Similarity search - Function | ||||||
| Biological species |  Halobacterium salinarum (Halophile) Halobacterium salinarum (Halophile) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.7 Å | ||||||
 Authors Authors | Luecke, H. / Schobert, B. / Cartailler, J.P. / Richter, H.T. / Rosengarth, A. / Needleman, R. / Lanyi, J.K. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: Coupling photoisomerization of retinal to directional transport in bacteriorhodopsin. Authors: Luecke, H. / Schobert, B. / Cartailler, J.P. / Richter, H.T. / Rosengarth, A. / Needleman, R. / Lanyi, J.K. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1f50.cif.gz | 69.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1f50.ent.gz | 48.9 KB | Display | PDB format |
| PDBx/mmJSON format | 1f50.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/f5/1f50ftp://data.pdbj.org/pub/pdb/validation_reports/f5/1f50 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1f4zC  1c3wS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |












-Links
 PDBj
PDBj





- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 24860.438 Da / Num. of mol.: 1 / Mutation: E204Q Source method: isolated from a genetically manipulated source Source: (gene. exp.) Halobacterium salinarum (Halophile) / Production host: Halobacterium salinarum (Halophile) / References: UniProt: P02945 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical | ChemComp-LI1 / 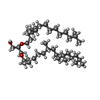  Mass: 639.130 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C42H86O3 Mass: 639.130 Da / Num. of mol.: 13 / Source method: obtained synthetically / Formula: C42H86O3#3: Chemical | ChemComp-SQU / |   Mass: 380.734 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H56 Mass: 380.734 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C27H56#4: Chemical | ChemComp-RET / |   Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O Mass: 284.436 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C20H28O#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 24 / Source method: isolated from a natural source / Formula: H2OHas protein modification | Y | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.31 Å3/Da / Density % sol: 46.7 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 295 K / Method: cubic lipid phase / pH: 5.6 Details: MO:WATER:PHOSPHATE, pH 5.6, Cubic lipid phase, temperature 295K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 20 ℃ / Method: batch methodDetails: Landau, E.M., (1996) Proc.Natl.Acad.Sci.USA., 93, 14532. | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| ||||||||||||||||||||
| Detector |
| ||||||||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | ||||||||||||||||||||
| Radiation wavelength | Wavelength: 1 Å / Relative weight: 1 | ||||||||||||||||||||
| Reflection | Resolution: 1.7→25 Å / Num. all: 257539 / Num. obs: 25709 / % possible obs: 99.1 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 10 % / Rmerge(I) obs: 0.057 / Net I/σ(I): 29.6 | ||||||||||||||||||||
| Reflection shell | Resolution: 1.7→1.73 Å / % possible obs: 86.4 % / Rmerge(I) obs: 0.296 / Mean I/σ(I) obs: 2.9 | ||||||||||||||||||||
| Reflection | *PLUS Num. measured all: 257539 |



- Processing
Processing
| Software |
| |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Starting model: ISOMORPHOUS TO 1C3W model Resolution: 1.7→12 Å / Num. parameters: 8300 / Num. restraintsaints: 8217 / Cross valid method: 5 / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber Details: Twin ratio is 50:50, R-factor (F>4sig)=0.120, R-free (F>4sig)=0.177
| |||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→12 Å
| |||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | |||||||||||||||||||||||||
| Refine LS restraints | *PLUS Type: s_bond_d / Dev ideal: 0.009 |
