 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1eex: CRYSTAL STRUCTURE OF THE DIOL DEHYDRATASE-ADENINYLPENTYLCOBALAMIN... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1eex | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF THE DIOL DEHYDRATASE-ADENINYLPENTYLCOBALAMIN COMPLEX FROM KLEBSIELLA OXYTOCA | ||||||
 Components Components | (PROPANEDIOL DEHYDRATASE) x 3 | ||||||
 Keywords Keywords | LYASE / COENZYME B12 / PROPANEDIOL / POTASSIUM ION / TIM BARREL | ||||||
| Function / homology |  Function and homology information Function and homology informationpropanediol dehydratase / propanediol dehydratase activity / cobalamin binding / metal ion binding Similarity search - Function | ||||||
| Biological species |  Klebsiella oxytoca (bacteria) Klebsiella oxytoca (bacteria) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.7 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.7 Å | ||||||
 Authors Authors | Shibata, N. / Masuda, J. / Toraya, T. / Morimoto, Y. / Yasuoka, N. | ||||||
 Citation Citation | Journal: Structure Fold.Des. / Year: 2000 Title: How a protein generates a catalytic radical from coenzyme B(12): X-ray structure of a diol-dehydratase-adeninylpentylcobalamin complex. Authors: Masuda, J. / Shibata, N. / Morimoto, Y. / Toraya, T. / Yasuoka, N. #1: Journal: Structure / Year: 1999Title: A new mode of B12 binding and the direct participation of a potassium ion in enzyme catalysis: X-ray structure of diol dehydratase Authors: Shibata, N. / Masuda, J. / Toraya, T. / Morimoto, Y. / Yasuoka, N. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1eex.cif.gz | 401.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1eex.ent.gz | 316.5 KB | Display | PDB format |
| PDBx/mmJSON format | 1eex.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ee/1eexftp://data.pdbj.org/pub/pdb/validation_reports/ee/1eex | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
| ||||||||
| Details | The protein exists as a dimer of heterotrimers. |
-Components
-Protein , 3 types, 6 molecules ALBEGM
| #1: Protein | Mass: 60408.133 Da / Num. of mol.: 2 / Fragment: ALPHA CHAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Klebsiella oxytoca (bacteria) / Plasmid: PUC119 / Production host: #2: Protein | Mass: 24141.678 Da / Num. of mol.: 2 / Fragment: BETA CHAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Klebsiella oxytoca (bacteria) / Plasmid: PUC119 / Production host: #3: Protein | Mass: 19198.695 Da / Num. of mol.: 2 / Fragment: GAMMA CHAIN Source method: isolated from a genetically manipulated source Source: (gene. exp.) Klebsiella oxytoca (bacteria) / Plasmid: PUC119 / Production host: |
|---|
-Non-polymers , 4 types, 1880 molecules 
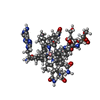
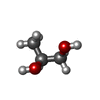

| #4: Chemical | ChemComp-K /  Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K Mass: 39.098 Da / Num. of mol.: 4 / Source method: obtained synthetically / Formula: K#5: Chemical |  Mass: 1537.631 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C72H106CoN18O14P Mass: 1537.631 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C72H106CoN18O14PDetails: ADENINYLPENTYLCOBALAMIN IS AN INACTIVE ANALOGUE OF ADENOSYLCOBALAMIN Adeninylpentylcobalamin is an inactive analogue of adenosylcobalamin. #6: Chemical |  Mass: 76.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2 Mass: 76.094 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C3H8O2#7: Water | ChemComp-HOH / | Mass: 18.015 Da / Num. of mol.: 1872 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 2 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.23 Å3/Da / Density % sol: 44.77 % | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, sandwich drop / pH: 8 Details: PEG 6000, potassium phospate, Tris-HCl, LDAO, ammonium sulfate, propane diol, adeninylpentylcobalamin,, pH 8.0, vapor diffusion, sandwich drop, temperature 277K | |||||||||||||||||||||||||
| Crystal grow | *PLUS Details: Masuda, J., (1999) Acta Crystallogr, D55, 907. | |||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Diffraction source |
| |||||||||||||||
| Detector |
| |||||||||||||||
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray | |||||||||||||||
| Radiation wavelength | Wavelength: 0.7 Å / Relative weight: 1 | |||||||||||||||
| Reflection | Resolution: 1.7→100 Å / Num. all: 206373 / Num. obs: 177514 / % possible obs: 86 % / Observed criterion σ(F): 1 / Observed criterion σ(I): 1 / Redundancy: 9.49 % / Biso Wilson estimate: 14.1 Å2 / Rmerge(I) obs: 0.079 / Net I/σ(I): 17.8 | |||||||||||||||
| Reflection shell | Resolution: 1.7→1.76 Å / Redundancy: 4.83 % / Rmerge(I) obs: 0.171 / Num. unique all: 12631 / % possible all: 61.8 | |||||||||||||||
| Reflection | *PLUS Num. measured all: 4543847 | |||||||||||||||
| Reflection shell | *PLUS % possible obs: 61.8 % |

- Processing
Processing
| Software |
| ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.7→30 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: Engh & Huber
| ||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.7→30 Å
| ||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||
| Software | *PLUS Name: SHELXL-97 / Classification: refinement | ||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 1.7 Å / Lowest resolution: 30 Å / σ(F): 0 | ||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||
| Displacement parameters | *PLUS |
