 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1cza: MUTANT MONOMER OF RECOMBINANT HUMAN HEXOKINASE TYPE I COMPLEXED W... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1cza | ||||||
|---|---|---|---|---|---|---|---|
| Title | MUTANT MONOMER OF RECOMBINANT HUMAN HEXOKINASE TYPE I COMPLEXED WITH GLUCOSE, GLUCOSE-6-PHOSPHATE, AND ADP | ||||||
 Components Components | HEXOKINASE TYPE I | ||||||
 Keywords Keywords | TRANSFERASE / STRUCTURALLY HOMOLOGOUS DOMAINS | ||||||
| Function / homology |  Function and homology information Function and homology informationDefective HK1 causes hexokinase deficiency (HK deficiency) / glucosamine kinase activity / : / hexokinase activity / Synthesis of GDP-mannose / carbohydrate phosphorylation / mannokinase activity / maintenance of protein location in mitochondrion / hexokinase / : ...Defective HK1 causes hexokinase deficiency (HK deficiency) / glucosamine kinase activity / : / hexokinase activity / Synthesis of GDP-mannose / carbohydrate phosphorylation / mannokinase activity / maintenance of protein location in mitochondrion / hexokinase / : / GDP-mannose biosynthetic process / fructokinase activity / glucokinase activity / mannose metabolic process / positive regulation of cytokine production involved in immune response / glucose 6-phosphate metabolic process / peptidoglycan binding / D-glucose binding / fructose 6-phosphate metabolic process / Glycolysis / canonical glycolysis / intracellular glucose homeostasis / glycolytic process / positive regulation of interleukin-1 beta production / glucose metabolic process / mitochondrial outer membrane / membrane raft / inflammatory response / innate immune response / mitochondrion / ATP binding / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.9 Å X-RAY DIFFRACTION / SYNCHROTRON / Resolution: 1.9 Å | ||||||
 Authors Authors | Aleshin, A.E. / Liu, X. / Kirby, C. / Bourenkov, G.P. / Bartunik, H.D. / Fromm, H.J. / Honzatko, R.B. | ||||||
 Citation Citation | Journal: J.Mol.Biol. / Year: 2000 Title: Crystal structures of mutant monomeric hexokinase I reveal multiple ADP binding sites and conformational changes relevant to allosteric regulation. Authors: Aleshin, A.E. / Kirby, C. / Liu, X. / Bourenkov, G.P. / Bartunik, H.D. / Fromm, H.J. / Honzatko, R.B. #1: Journal: J.Mol.Biol. / Year: 1998Title: Regulation of Hexokinase I: Crystal Structure of Recombinant Human Brain Hexokinase Complexed with Glucose and Phosphate Authors: Aleshin, A.E. / Zeng, C. / Bartunik, H.D. / Fromm, H.J. / Honzatko, R.B. #2: Journal: Structure / Year: 1998Title: The Mechanism of Regulation of Hexokinase: New Insights from the Crystal Structure of Recombinant Human Brain Hexokinase Complexed with Glucose and Glucose-6-Phosphate Authors: Aleshin, A.E. / Zeng, C. / Bourenkov, G.P. / Bartunik, H.D. / Fromm, H.J. / Honzatko, R.B. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1cza.cif.gz | 208.3 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1cza.ent.gz | 163 KB | Display | PDB format |
| PDBx/mmJSON format | 1cza.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/cz/1czaftp://data.pdbj.org/pub/pdb/validation_reports/cz/1cza | HTTPS FTP |
|---|
-Related structure data













-Links
 PDBj
PDBj


- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 102587.008 Da / Num. of mol.: 1 / Mutation: E280A, R283A, G284Y Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cell: NEURON / Organ: BRAIN / Plasmid: PET11A / Production host:  | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Sugar | 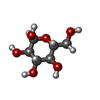  Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H12O6 Type: D-saccharide, alpha linking / Mass: 180.156 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H12O6#3: Sugar | 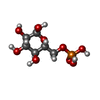  Type: D-saccharide, alpha linking / Mass: 260.136 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13O9P Type: D-saccharide, alpha linking / Mass: 260.136 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C6H13O9P#4: Chemical | ChemComp-ADP / |   Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM Mass: 427.201 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C10H15N5O10P2 / Comment: ADP, energy-carrying molecule*YM#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 672 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 672 / Source method: isolated from a natural source / Formula: H2OCompound details | WILD-TYPE HEXOKINASE I IS A MONOMER AT CONCENTRATION BELOW 1 MG/ML. THE MUTANT IN THIS ENTRY IS A ...WILD-TYPE HEXOKINASE | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.01 Å3/Da / Density % sol: 59.11 % | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: evaporation / pH: 6.5 Details: PEG 8000, SODIUM ACETATE, MES, ADP, ALUMINUM NITRATE, SODIUM FLUORIDE, MAGNESIUM ACETATE, GLUCOSE, pH 6.5, EVAPORATION, temperature 298.0K | ||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 7.5 / Method: unknown | ||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 110 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: EMBL/DESY, HAMBURG  / Beamline: BW7B / Wavelength: 1.05 / Beamline: BW7B / Wavelength: 1.05 |
| Detector | Type: MARRESEARCH / Detector: CCD / Date: Sep 6, 1998 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.05 Å / Relative weight: 1 |
| Reflection | Resolution: 1.9→17 Å / Num. all: 96631 / Num. obs: 96631 / % possible obs: 98.3 % / Observed criterion σ(F): 0 / Observed criterion σ(I): 0 / Redundancy: 4.7 % / Biso Wilson estimate: 30.6 Å2 / Rmerge(I) obs: 0.055 / Net I/σ(I): 16.8 |
| Reflection shell | Resolution: 1.9→1.97 Å / Redundancy: 4 % / Rmerge(I) obs: 0.55 / % possible all: 98.3 |
| Reflection | *PLUS Num. obs: 83349 / % possible obs: 98.6 % / Num. measured all: 286697 / Rmerge(I) obs: 0.048 |
| Reflection shell | *PLUS % possible obs: 82.3 % / Rmerge(I) obs: 0.41 |
- Processing
Processing
| Software |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.9→8 Å / σ(F): 0 / σ(I): 0 / Stereochemistry target values: ENGH & HUBER Details: USED MAXIMUM LIKELIHOOD RESIDUALS AND CONJUGATE DIRECTION MINIMIZATION ADP is bound near the N-terminus of the polypeptide chain. Side chains of residues 16, 20, 24, 101, 102, 353, 794, 801, ...Details: USED MAXIMUM LIKELIHOOD RESIDUALS AND CONJUGATE DIRECTION MINIMIZATION ADP is bound near the N-terminus of the polypeptide chain. Side chains of residues 16, 20, 24, 101, 102, 353, 794, 801, 811 have poor electron density. Their occupancies are set to 0.01.
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.9→8 Å
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Highest resolution: 2 Å / Rfactor Rfree: 0.268 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
