 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1bg3: RAT BRAIN HEXOKINASE TYPE I COMPLEX WITH GLUCOSE AND INHIBITOR GL... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1bg3 | ||||||
|---|---|---|---|---|---|---|---|
| Title | RAT BRAIN HEXOKINASE TYPE I COMPLEX WITH GLUCOSE AND INHIBITOR GLUCOSE-6-PHOSPHATE | ||||||
 Components Components | HEXOKINASE | ||||||
 Keywords Keywords | HEXOKINASE / PHOSPHOTRANSFERASE | ||||||
| Function / homology |  Function and homology information Function and homology informationnegative regulation of anion transmembrane transport / response to ketamine / glucosamine kinase activity / hexokinase activity / response to brassinosteroid / carbohydrate phosphorylation / mannokinase activity / maintenance of protein location in mitochondrion / hexokinase / : ...negative regulation of anion transmembrane transport / response to ketamine / glucosamine kinase activity / hexokinase activity / response to brassinosteroid / carbohydrate phosphorylation / mannokinase activity / maintenance of protein location in mitochondrion / hexokinase / : / fructokinase activity / glucokinase activity / mannose metabolic process / positive regulation of cytokine production involved in immune response / glucose 6-phosphate metabolic process / peptidoglycan binding / D-glucose binding / fructose 6-phosphate metabolic process / canonical glycolysis / peptidyl-threonine phosphorylation / peptidyl-tyrosine phosphorylation / response to ischemia / intracellular glucose homeostasis / glycolytic process / positive regulation of interleukin-1 beta production / peptidyl-serine phosphorylation / sperm principal piece / glucose metabolic process / caveola / protein autophosphorylation / cilium / response to hypoxia / protein phosphorylation / protein kinase activity / mitochondrial outer membrane / inflammatory response / membrane raft / innate immune response / negative regulation of apoptotic process / ATP hydrolysis activity / protein-containing complex / mitochondrion / ATP binding / identical protein binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.8 Å | ||||||
 Authors Authors | Mulichak, A.M. / Garavito, R.M. | ||||||
 Citation Citation | Journal: Nat.Struct.Biol. / Year: 1998 Title: The structure of mammalian hexokinase-1. Authors: Mulichak, A.M. / Wilson, J.E. / Padmanabhan, K. / Garavito, R.M. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1bg3.cif.gz | 351 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1bg3.ent.gz | 281.1 KB | Display | PDB format |
| PDBx/mmJSON format | 1bg3.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/bg/1bg3ftp://data.pdbj.org/pub/pdb/validation_reports/bg/1bg3 | HTTPS FTP |
|---|
-Related structure data









-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (-0.999992, 0.001479, -0.003776), Vector: Details | ALTHOUGH ENZYME IS ACTIVE AS A MONOMER, DIMERIZATION OCCURS AT HIGH PROTEIN CONCENTRATION, PARTICULARLY IN THE PRESENCE OF THE INHIBITOR G6P | |
-Components
| #1: Protein | Mass: 102545.156 Da / Num. of mol.: 2 / Source method: isolated from a natural source Details: BOTH MONOMERS HAVE TWO SMALL BREAKS AT KNOWN OR LIKELY TRYPSIN CLEAVAGE SITES. ALTHOUGH ENZYME IS ACTIVE AS A MONOMER, DIMERIZATION OCCURS AT HIGH PROTEIN CONCENTRATION, PARTICULARLY IN THE ...Details: BOTH MONOMERS HAVE TWO SMALL BREAKS AT KNOWN OR LIKELY TRYPSIN CLEAVAGE SITES. ALTHOUGH ENZYME IS ACTIVE AS A MONOMER, DIMERIZATION OCCURS AT HIGH PROTEIN CONCENTRATION, PARTICULARLY IN THE PRESENCE OF THE INHIBITOR G6P Source: (natural) #2: Sugar | ChemComp-BGC / 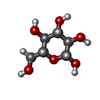  Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 4 Type: D-saccharide, beta linking / Mass: 180.156 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H12O6 #3: Sugar | ChemComp-G6P / 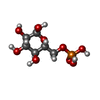  Type: D-saccharide, alpha linking / Mass: 260.136 Da / Num. of mol.: 4 Type: D-saccharide, alpha linking / Mass: 260.136 Da / Num. of mol.: 4Source method: isolated from a genetically manipulated source Formula: C6H13O9P #4: Chemical | ChemComp-CA / |   Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca Mass: 40.078 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Ca#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 234 / Source method: isolated from a natural source / Formula: H2OCompound details | BOTH MONOMERS HAVE TWO SMALL BREAKS AT KNOWN OR LIKELY TRYPSIN CLEAVAGE SITES. | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 3.5 Å3/Da / Density % sol: 55 % | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 7 / Details: pH 7.0 | ||||||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 153 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS II / Detector: IMAGE PLATE / Date: Aug 1, 1997 / Details: MSC FOCUSSING MIRRORS |
| Radiation | Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Highest resolution: 2.8 Å / Num. obs: 54454 / % possible obs: 79 % / Observed criterion σ(I): 1 / Redundancy: 2 % / Rmerge(I) obs: 0.076 / Net I/σ(I): 8 |
| Reflection shell | Resolution: 2.8→3 Å / Rmerge(I) obs: 0.17 / Mean I/σ(I) obs: 3 / % possible all: 59 |
| Reflection shell | *PLUS % possible obs: 59 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: HEXOKINASE FROM SCHISTOSOMA MANSONI Resolution: 2.8→20 Å / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 20 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.8→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.8→2.9 Å / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.1 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
|
