 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1ci1: CRYSTAL STRUCTURE OF TRIOSEPHOSPHATE ISOMERASE FROM TRYPANOSOMA C... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1ci1 | ||||||
|---|---|---|---|---|---|---|---|
| Title | CRYSTAL STRUCTURE OF TRIOSEPHOSPHATE ISOMERASE FROM TRYPANOSOMA CRUZI IN HEXANE | ||||||
 Components Components | PROTEIN (TRIOSEPHOSPHATE ISOMERASE) | ||||||
 Keywords Keywords | TRIOSEPHOSPHATE ISOMERASE / TRYPANOSOMA CRUZI / ORGANIC SOLVENT / HEXANE / OLIGOMERIC PROTEIN | ||||||
| Function / homology |  Function and homology information Function and homology informationglycosome / triose-phosphate isomerase / triose-phosphate isomerase activity / glyceraldehyde-3-phosphate biosynthetic process / glycerol catabolic process / glycolytic process / gluconeogenesis / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / OTHER / Resolution: 2 Å X-RAY DIFFRACTION / OTHER / Resolution: 2 Å | ||||||
 Authors Authors | Gao, X.-G. / Maldondo, E. / Perez-Montfort, R. / De Gomez-Puyou, M.T. / Gomez-Puyou, A. / Rodriguez-Romero, A. | ||||||
 Citation Citation | Journal: Proc.Natl.Acad.Sci.USA / Year: 1999 Title: Crystal structure of triosephosphate isomerase from Trypanosoma cruzi in hexane. Authors: Gao, X.G. / Maldonado, E. / Perez-Montfort, R. / Garza-Ramos, G. / de Gomez-Puyou, M.T. / Gomez-Puyou, A. / Rodriguez-Romero, A. #1: Journal: J.Mol.Biol. / Year: 1998Title: Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes. Authors: Maldonado, E. / Soriano-Garcia, M. / Moreno, A. / Cabrera, N. / Garza-Ramos, G. / de Gomez-Puyou, M. / Gomez-Puyou, A. / Perez-Montfort, R. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1ci1.cif.gz | 110.8 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1ci1.ent.gz | 85.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1ci1.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1ci1_validation.pdf.gz | 431.1 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1ci1_full_validation.pdf.gz | 438.3 KB | Display | |
| Data in XML | 1ci1_validation.xml.gz | 21.9 KB | Display | |
| Data in CIF | 1ci1_validation.cif.gz | 31.2 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/ci/1ci1ftp://data.pdbj.org/pub/pdb/validation_reports/ci/1ci1 | HTTPS FTP |
-Related structure data
| Related structure data |  1tcdS S: Starting model for refinement |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 27360.438 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.)  #2: Chemical | 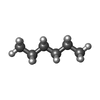  Mass: 86.175 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14 Mass: 86.175 Da / Num. of mol.: 3 / Source method: obtained synthetically / Formula: C6H14#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 236 / Source method: isolated from a natural source / Formula: H2OSequence details | THE FIRST 3 N-TERMINAL RESIDUE WERE NOT BUILT INTO THE MODEL A AS THEY WERE NOT SEEEN IN THE ...THE FIRST 3 N-TERMINAL RESIDUE WERE NOT BUILT INTO THE MODEL A AS THEY WERE NOT SEEEN IN THE DENSITY MAPS THE FIRST 2 N-TERMINAL RESIDUE WERE NOT BUILT INTO THE MODEL B AS THEY WERE NOT SEEN IN THE DENSITY MAPS | |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.32 Å3/Da / Density % sol: 47 % | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Method: vapor diffusion / pH: 7.5 Details: PROTEIN WAS CRYSTALLIZED AT ROOM TEMPERATUTE BY VAPER DIFFUSION FROM 0.1 M NA HEPES PH7.5, 2%(V/V) PEG400 AND 2.0 M AMMONIUM SULFATE, THEN SOAKED IN ANHYDROUS N-HEXANE. , VAPOR DIFFUSION | ||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||
| Crystal grow | *PLUS Method: vapor diffusion, hanging drop | ||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 291 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RU200 / Wavelength: 1.5418 |
| Detector | Type: RIGAKU RAXIS IIC / Detector: IMAGE PLATE / Date: Apr 21, 1998 / Details: MIRRORS |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 1.95→25 Å / Num. obs: 31766 / % possible obs: 84.4 % / Observed criterion σ(I): 2 / Redundancy: 3.6 % / Biso Wilson estimate: 18.1 Å2 / Rmerge(I) obs: 0.116 / Net I/σ(I): 4.64 |
| Reflection shell | Resolution: 2→2.11 Å / Redundancy: 3.4 % / Rmerge(I) obs: 0.113 / Mean I/σ(I) obs: 2.1 / % possible all: 65.5 |
| Reflection | *PLUS Num. measured all: 172135 |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: OTHER Starting model: PDB ENTRY 1TCD Resolution: 2→5 Å / Rfactor Rfree error: 0.005 / Data cutoff high absF: 10000000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: THROUGHOUT / σ(F): 2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 25.6 Å2
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2→5 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2→2.12 Å / Rfactor Rfree error: 0.019 / Total num. of bins used: 6
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3.851 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Lowest resolution: 5 Å / σ(F): 2 / % reflection Rfree: 10 % / Rfactor obs: 0.181 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS Biso mean: 25.6 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor Rfree: 0.358 / % reflection Rfree: 9.9 % / Rfactor Rwork: 0.309 |
