 Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1c22 | ||||||
|---|---|---|---|---|---|---|---|
| Title | E. COLI METHIONINE AMINOPEPTIDASE: TRIFLUOROMETHIONINE COMPLEX | ||||||
 Components Components | METHIONINE AMINOPEPTIDASE | ||||||
 Keywords Keywords | HYDROLASE / PRODUCT COMPLEX | ||||||
| Function / homology |  Function and homology information Function and homology information: / methionyl aminopeptidase / initiator methionyl aminopeptidase activity / metalloaminopeptidase activity / ferrous iron binding / proteolysis / metal ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / Resolution: 1.75 Å X-RAY DIFFRACTION / Resolution: 1.75 Å | ||||||
 Authors Authors | Lowther, W.T. / Zhang, Y. / Sampson, P.B. / Honek, J.F. / Matthews, B.W. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1999 Title: Insights into the mechanism of Escherichia coli methionine aminopeptidase from the structural analysis of reaction products and phosphorus-based transition-state analogues. Authors: Lowther, W.T. / Zhang, Y. / Sampson, P.B. / Honek, J.F. / Matthews, B.W. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1c22.cif.gz | 67 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1c22.ent.gz | 47.8 KB | Display | PDB format |
| PDBx/mmJSON format | 1c22.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/c2/1c22ftp://data.pdbj.org/pub/pdb/validation_reports/c2/1c22 | HTTPS FTP |
|---|
-Related structure data























-Links
 PDBj
PDBj
- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 |
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 29210.580 Da / Num. of mol.: 1 / Mutation: R175Q Source method: isolated from a genetically manipulated source Details: TRIFLUOROMETHIONINE COMPLEX / Source: (gene. exp.) References: UniProt: P07906, UniProt: P0AE18*PLUS, methionyl aminopeptidase | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| #2: Chemical |   Mass: 58.933 Da / Num. of mol.: 2 / Fragment: TRIFLUOROMETHIONINE / Source method: obtained synthetically / Formula: Co Mass: 58.933 Da / Num. of mol.: 2 / Fragment: TRIFLUOROMETHIONINE / Source method: obtained synthetically / Formula: Co#3: Chemical | ChemComp-NA / |   Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na Mass: 22.990 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: Na#4: Chemical | ChemComp-MF3 / | 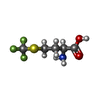  Type: L-peptide linking / Mass: 203.183 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H8F3NO2S Type: L-peptide linking / Mass: 203.183 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C5H8F3NO2S#5: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 103 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 103 / Source method: isolated from a natural source / Formula: H2OSequence details | THERE ARE FOUR ADDITIONAL RESIDUES ON THE C-TERMINUS LEFT OVER FROM A THROMBIN DIGEST OF THE HIS- ...THERE ARE FOUR ADDITIONAL | |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.06 Å3/Da / Density % sol: 40.36 % | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 298 K / Method: vapor diffusion, sitting drop Details: HEPES, CoCl2, K2SO4, Methionine, PEG4000, NaCl, N-octanoyl sucrose, VAPOR DIFFUSION, SITTING DROP, temperature 298K | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS pH: 6.8 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 298 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU / Wavelength: 1.54 |
| Detector | Type: RIGAKU RAXIS / Detector: IMAGE PLATE / Date: Mar 18, 1999 |
| Radiation | Protocol: SINGLE WAVELENGTH / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.54 Å / Relative weight: 1 |
| Reflection | Resolution: 1.75→27.1 Å / Num. all: 24021 / Num. obs: 116107 / % possible obs: 99.9 % / Redundancy: 4.8 % / Biso Wilson estimate: 17.2 Å2 / Rmerge(I) obs: 0.049 / Net I/σ(I): 35.2 |
| Reflection shell | Resolution: 1.75→1.81 Å / Rmerge(I) obs: 0.264 / Mean I/σ(I) obs: 4.5 / % possible all: 100 |
| Reflection | *PLUS Num. obs: 24021 / Num. measured all: 116107 |
| Reflection shell | *PLUS % possible obs: 100 % |
- Processing
Processing
| Software |
| |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Resolution: 1.75→27.1 Å / Stereochemistry target values: TNT /
| |||||||||||||||
| Solvent computation | Solvent model: TNT / Bsol: 221.4 Å2 / ksol: 0.842 e/Å3 | |||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→27.1 Å
| |||||||||||||||
| Refine LS restraints |
| |||||||||||||||
| Software | *PLUS Name: TNT / Classification: refinement | |||||||||||||||
| Refinement | *PLUS Num. reflection obs: 24021 / Rfactor all: 0.163 | |||||||||||||||
| Solvent computation | *PLUS | |||||||||||||||
| Displacement parameters | *PLUS | |||||||||||||||
| Refine LS restraints | *PLUS
|
