 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1beh: HUMAN PHOSPHATIDYLETHANOLAMINE BINDING PROTEIN IN COMPLEX WITH CA... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1beh | ||||||
|---|---|---|---|---|---|---|---|
| Title | HUMAN PHOSPHATIDYLETHANOLAMINE BINDING PROTEIN IN COMPLEX WITH CACODYLATE | ||||||
 Components Components | PHOSPHATIDYLETHANOLAMINE BINDING PROTEIN | ||||||
 Keywords Keywords | LIPID BINDING PROTEIN / LIPID-BINDING / SIGNALLING | ||||||
| Function / homology |  Function and homology information Function and homology informationphosphatidylethanolamine binding / negative regulation of MAPK cascade / serine-type endopeptidase inhibitor activity / Signaling by high-kinase activity BRAF mutants / MAP2K and MAPK activation / Negative regulation of MAPK pathway / Signaling by moderate kinase activity BRAF mutants / Paradoxical activation of RAF signaling by kinase inactive BRAF / Signaling downstream of RAS mutants / Signaling by BRAF and RAF1 fusions ...phosphatidylethanolamine binding / negative regulation of MAPK cascade / serine-type endopeptidase inhibitor activity / Signaling by high-kinase activity BRAF mutants / MAP2K and MAPK activation / Negative regulation of MAPK pathway / Signaling by moderate kinase activity BRAF mutants / Paradoxical activation of RAF signaling by kinase inactive BRAF / Signaling downstream of RAS mutants / Signaling by BRAF and RAF1 fusions / protein kinase binding / enzyme binding / RNA binding / extracellular exosome / ATP binding / nucleus / cytosol Similarity search - Function | ||||||
| Biological species |  Homo sapiens (human) Homo sapiens (human) | ||||||
| Method |  X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å X-RAY DIFFRACTION / SYNCHROTRON / MOLECULAR REPLACEMENT / Resolution: 1.75 Å | ||||||
 Authors Authors | Banfield, M.J. / Barker, J.J. / Perry, A. / Brady, R.L. | ||||||
 Citation Citation | Journal: Structure / Year: 1998 Title: Function from structure? The crystal structure of human phosphatidylethanolamine-binding protein suggests a role in membrane signal transduction. Authors: Banfield, M.J. / Barker, J.J. / Perry, A.C. / Brady, R.L. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1beh.cif.gz | 88.1 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1beh.ent.gz | 67.2 KB | Display | PDB format |
| PDBx/mmJSON format | 1beh.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Summary document | 1beh_validation.pdf.gz | 445.6 KB | Display | wwPDB validaton report |
|---|---|---|---|---|
| Full document | 1beh_full_validation.pdf.gz | 450.4 KB | Display | |
| Data in XML | 1beh_validation.xml.gz | 18.3 KB | Display | |
| Data in CIF | 1beh_validation.cif.gz | 26.1 KB | Display | |
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/be/1behftp://data.pdbj.org/pub/pdb/validation_reports/be/1beh | HTTPS FTP |
-Related structure data
| Related structure data |  1bd9SC S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |






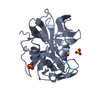



-Links
 PDBj
PDBj



- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 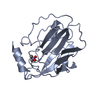
| ||||||||
| 2 | 
| ||||||||
| Unit cell |
| ||||||||
| Noncrystallographic symmetry (NCS) | NCS oper: (Code: given Matrix: (0.48316, 0.87203, -0.07827), Vector: |
-Components
| #1: Protein | Mass: 21086.787 Da / Num. of mol.: 2 Source method: isolated from a genetically manipulated source Source: (gene. exp.) Homo sapiens (human) / Cellular location: CYTOPLASM, MEMBRANE-ASSOCIATED / Plasmid: PET-15B / Species (production host): Escherichia coli / Cellular location (production host): CYTOPLASM / Production host:  #2: Chemical | 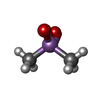  Mass: 136.989 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6AsO2 Mass: 136.989 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: C2H6AsO2#3: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 239 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 239 / Source method: isolated from a natural source / Formula: H2O |
|---|
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 1 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.2 Å3/Da / Density % sol: 36 % | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | pH: 6.5 Details: PROTEIN WAS CRYSTALLIZED FROM 28-32% PEG 4000/6000/8000, 200-300 MM SODIUM ACETATE, 100MM SODIUM CACODYLATE., pH 6.5 | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Method: unknown | ||||||||||||||||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 100 K |
|---|---|
| Diffraction source | Source: SYNCHROTRON / Site: SRS  / Beamline: PX7.2 / Wavelength: 1.488 / Beamline: PX7.2 / Wavelength: 1.488 |
| Detector | Type: MARRESEARCH / Detector: IMAGE PLATE / Date: Apr 15, 1997 / Details: MIRRORS |
| Radiation | Monochromator: GE(111) / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.488 Å / Relative weight: 1 |
| Reflection | Resolution: 1.7→20 Å / Num. obs: 38803 / % possible obs: 95.6 % / Observed criterion σ(I): -3 / Redundancy: 2.4 % / Biso Wilson estimate: 19.1 Å2 / Rmerge(I) obs: 0.055 / Net I/σ(I): 16.2 |
| Reflection shell | Resolution: 1.7→1.76 Å / Redundancy: 2.3 % / Rmerge(I) obs: 0.28 / Mean I/σ(I) obs: 2.6 / % possible all: 94.9 |
| Reflection shell | *PLUS % possible obs: 94.9 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 1BD9 Resolution: 1.75→20 Å / Cross valid method: THROUGHOUT / σ(F): 0
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 24 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 1.75→20 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: REFMAC / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS Rfactor obs: 0.235 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Highest resolution: 1.75 Å / Lowest resolution: 1.83 Å / Rfactor all: 0.268 / Rfactor obs: 0.322 |
