 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- PDB-1a72: AN ACTIVE-SITE DOUBLE MUTANT (PHE93->TRP, VAL203->ALA) OF HORSE L... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: PDB / ID: 1a72 | ||||||
|---|---|---|---|---|---|---|---|
| Title | AN ACTIVE-SITE DOUBLE MUTANT (PHE93->TRP, VAL203->ALA) OF HORSE LIVER ALCOHOL DEHYDROGENASE IN COMPLEX WITH THE ISOSTERIC NAD ANALOG CPAD | ||||||
 Components Components | HORSE LIVER ALCOHOL DEHYDROGENASE | ||||||
 Keywords Keywords | OXIDOREDUCTASE / OXIDOREDUCTASE (NAD(A)-CHOH(D)) / ACTIVE SITE MUTANT / LIVER ALCOHOL DEHYDROGENASE / ISOSTERIC NAD INHIBITORS | ||||||
| Function / homology |  Function and homology information Function and homology informationall-trans-retinol dehydrogenase (NAD+) activity / alcohol dehydrogenase / retinoic acid metabolic process / retinol metabolic process / zinc ion binding / cytosol Similarity search - Function | ||||||
| Biological species |  | ||||||
| Method |  X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å X-RAY DIFFRACTION / MOLECULAR REPLACEMENT / Resolution: 2.6 Å | ||||||
 Authors Authors | Colby, T.D. / Bahnson, B.J. / Chin, J.K. / Klinman, J.P. / Goldstein, B.M. | ||||||
 Citation Citation | Journal: Biochemistry / Year: 1998 Title: Active site modifications in a double mutant of liver alcohol dehydrogenase: structural studies of two enzyme-ligand complexes. Authors: Colby, T.D. / Bahnson, B.J. / Chin, J.K. / Klinman, J.P. / Goldstein, B.M. #1: Journal: Proc.Natl.Acad.Sci.USA / Year: 1997Title: A Link between Protein Structure and Enzyme Catalyzed Hydrogen Tunneling Authors: Bahnson, B.J. / Colby, T.D. / Chin, J.K. / Goldstein, B.M. / Klinman, J.P. | ||||||
| History |
|
- Structure visualization
Structure visualization
| Structure viewer | Molecule: MolmilJmol/JSmol |
|---|
- Downloads & links
Downloads & links
-Download
| PDBx/mmCIF format | 1a72.cif.gz | 85.4 KB | Display | PDBx/mmCIF format |
|---|---|---|---|---|
| PDB format | pdb1a72.ent.gz | 63.4 KB | Display | PDB format |
| PDBx/mmJSON format | 1a72.json.gz | Tree view | PDBx/mmJSON format | |
| Others |  Other downloads Other downloads |
-Validation report
| Arichive directory | https://data.pdbj.org/pub/pdb/validation_reports/a7/1a72ftp://data.pdbj.org/pub/pdb/validation_reports/a7/1a72 | HTTPS FTP |
|---|
-Related structure data
| Related structure data |  1a71C  8adhS S: Starting model for refinement C: citing same article ( |
|---|---|
| Similar structure data |




















-Links
 PDBj
PDBj

- Assembly
Assembly
| Deposited unit | 
| ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 
| ||||||||
| Unit cell |
|
-Components
| #1: Protein | Mass: 39864.258 Da / Num. of mol.: 1 / Mutation: F93W, V203A Source method: isolated from a genetically manipulated source Source: (gene. exp.)  | ||||
|---|---|---|---|---|---|
| #2: Chemical |   Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn Mass: 65.409 Da / Num. of mol.: 2 / Source method: obtained synthetically / Formula: Zn#3: Chemical | ChemComp-PAD / | 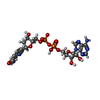  Mass: 663.425 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H27N7O14P2 Mass: 663.425 Da / Num. of mol.: 1 / Source method: obtained synthetically / Formula: C21H27N7O14P2#4: Water | ChemComp-HOH / |  Mass: 18.015 Da / Num. of mol.: 78 / Source method: isolated from a natural source / Formula: H2O Mass: 18.015 Da / Num. of mol.: 78 / Source method: isolated from a natural source / Formula: H2O |
-Experimental details
-Experiment
| Experiment | Method: X-RAY DIFFRACTION / Number of used crystals: 3 |
|---|
- Sample preparation
Sample preparation
| Crystal | Density Matthews: 2.3 Å3/Da / Density % sol: 46.82 % | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Crystal grow | Temperature: 277 K / Method: vapor diffusion, hanging drop / pH: 8.4 Details: CRYSTALS GROWN FROM 4 MICROLITER HANGING DROPS OF 10MG/ML PROTEIN IN 50 MM TRIS BUFFER (PH8.4 @4C) 4% (V/V) ETOH EQUILIBRATED AT 4C WITH WELLS CONTAINING 11-13% ETOH, vapor diffusion - ...Details: CRYSTALS GROWN FROM 4 MICROLITER HANGING DROPS OF 10MG/ML PROTEIN IN 50 MM TRIS BUFFER (PH8.4 @4C) 4% (V/V) ETOH EQUILIBRATED AT 4C WITH WELLS CONTAINING 11-13% ETOH, vapor diffusion - hanging drop, temperature 277K | |||||||||||||||||||||||||||||||||||
| Crystal grow | *PLUS Temperature: 4 ℃ / Method: vapor diffusion | |||||||||||||||||||||||||||||||||||
| Components of the solutions | *PLUS
|
-Data collection
| Diffraction | Mean temperature: 275 K |
|---|---|
| Diffraction source | Source: ROTATING ANODE / Type: RIGAKU RUH2R / Wavelength: 1.5418 |
| Detector | Type: XENTRONICS / Detector: AREA DETECTOR / Date: Dec 15, 1993 |
| Radiation | Monochromator: NI FILTER / Monochromatic (M) / Laue (L): M / Scattering type: x-ray |
| Radiation wavelength | Wavelength: 1.5418 Å / Relative weight: 1 |
| Reflection | Resolution: 2.6→22 Å / Num. obs: 10388 / % possible obs: 76.9 % / Observed criterion σ(I): 2 / Biso Wilson estimate: 38 Å2 / Rmerge(I) obs: 0.078 / Rsym value: 0.078 / Net I/σ(I): 6 |
| Reflection shell | Resolution: 2.56→2.68 Å / Mean I/σ(I) obs: 2 / % possible all: 43 |
| Reflection | *PLUS Num. measured all: 16939 / Rmerge(I) obs: 0.078 |
| Reflection shell | *PLUS % possible obs: 43 % |
- Processing
Processing
| Software |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Refinement | Method to determine structure: MOLECULAR REPLACEMENT Starting model: PDB ENTRY 8ADH Resolution: 2.6→8 Å / Rfactor Rfree error: 0.01 / Data cutoff high absF: 100000 / Data cutoff low absF: 0.001 / Isotropic thermal model: RESTRAINED / Cross valid method: A POSTERIORI / σ(F): 2 Details: RFREE FIGURE BELONGS TO SA OMIT PROCEDURE USED TO CONFIRM FIT OF LIGAND. THIS RFREE DESCRIBES A MODEL NOT CONTAINING SOLVENT
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | Biso mean: 16.7 Å2 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine analyze | Luzzati coordinate error obs: 0.36 Å / Luzzati d res low obs: 5 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement step | Cycle: LAST / Resolution: 2.6→8 Å
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | Resolution: 2.6→2.71 Å / Rfactor Rfree error: 0.04 / Total num. of bins used: 8
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Xplor file |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Software | *PLUS Name: X-PLOR / Version: 3 / Classification: refinement | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refinement | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Solvent computation | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Displacement parameters | *PLUS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Refine LS restraints | *PLUS
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| LS refinement shell | *PLUS Rfactor obs: 0.234 |
