National Institutes of Health/National Institute of General Medical Sciences (NIH/NIGMS)
R01-GM63072
United States
Citation
Journal: Nature / Year: 2017 Title: The cryo-electron microscopy structure of human transcription factor IIH. Authors: Basil J Greber / Thi Hoang Duong Nguyen / Jie Fang / Pavel V Afonine / Paul D Adams / Eva Nogales / Abstract: Human transcription factor IIH (TFIIH) is part of the general transcriptional machinery required by RNA polymerase II for the initiation of eukaryotic gene transcription. Composed of ten subunits ...Human transcription factor IIH (TFIIH) is part of the general transcriptional machinery required by RNA polymerase II for the initiation of eukaryotic gene transcription. Composed of ten subunits that add up to a molecular mass of about 500 kDa, TFIIH is also essential for nucleotide excision repair. The seven-subunit TFIIH core complex formed by XPB, XPD, p62, p52, p44, p34, and p8 is competent for DNA repair, while the CDK-activating kinase subcomplex, which includes the kinase activity of CDK7 as well as the cyclin H and MAT1 subunits, is additionally required for transcription initiation. Mutations in the TFIIH subunits XPB, XPD, and p8 lead to severe premature ageing and cancer propensity in the genetic diseases xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy, highlighting the importance of TFIIH for cellular physiology. Here we present the cryo-electron microscopy structure of human TFIIH at 4.4 Å resolution. The structure reveals the molecular architecture of the TFIIH core complex, the detailed structures of its constituent XPB and XPD ATPases, and how the core and kinase subcomplexes of TFIIH are connected. Additionally, our structure provides insight into the conformational dynamics of TFIIH and the regulation of its activity.
History
Deposition
Jul 10, 2017
-
Header (metadata) release
Jul 26, 2017
-
Map release
Oct 4, 2017
-
Update
Jan 29, 2020
-
Current status
Jan 29, 2020
Processing site: RCSB / Status: Released
-
Structure visualization
Movie
Surface view with section colored by density value
Organism: Homo sapiens (human) / Strain: HeLa / Organelle: Nucleus / Location in cell: Nucleus
Molecular weight
Theoretical: 490 KDa
-
Experimental details
-
Structure determination
Method
cryo EM
Processing
single particle reconstruction
Aggregation state
particle
-
Sample preparation
Concentration
0.0049 mg/mL
Buffer
pH: 7.8 Component:
Concentration
Name
Formula
20.0 mM
HEPES-KOH
150.0 mM
potassium chloride
KCl
5.0 mM
magnesium chloride
MgCl2
0.5 mM
DTT
2.0 %
trehalose
1.5 %
glycerol
0.015 %
NP-40 substitute
Grid
Model: Protochips C-flat 4/2 / Material: COPPER / Mesh: 400 / Support film - #0 - Film type ID: 1 / Support film - #0 - Material: CARBON / Support film - #0 - topology: HOLEY / Support film - #1 - Film type ID: 2 / Support film - #1 - Material: CARBON / Support film - #1 - topology: CONTINUOUS / Pretreatment - Type: PLASMA CLEANING / Pretreatment - Atmosphere: AIR
Vitrification
Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 278.15 K / Instrument: FEI VITROBOT MARK IV / Details: 3-4 minute incubation, 2 second blot.
Details
Natively purified complex at approx. 10 nM concentration
-
Electron microscopy
Microscope
FEI TITAN
Image recording
Film or detector model: GATAN K2 SUMMIT (4k x 4k) / Detector mode: COUNTING / Digitization - Dimensions - Width: 3840 pixel / Digitization - Dimensions - Height: 3710 pixel / Digitization - Sampling interval: 5.0 µm / Digitization - Frames/image: 1-30 / Number grids imaged: 4 / Number real images: 8300 / Average exposure time: 8.7 sec. / Average electron dose: 40.0 e/Å2 Details: 8300 micrographs collected in four session with identical acquisition settings. Sessions lasted 4, 2, 4, and 4 days and yielded 1200, 1700, 2800, and 2600 micrographs.
Electron beam
Acceleration voltage: 300 kV / Electron source: FIELD EMISSION GUN
Specimen holder model: GATAN 626 SINGLE TILT LIQUID NITROGEN CRYO TRANSFER HOLDER Cooling holder cryogen: NITROGEN
+
Image processing
Details
Micrographs were inspected for quality of Thon rings and ice contamination. Poor micrographs were rejected.
Particle selection
Number selected: 1500000 Details: Initial rounds of particle selection using DogPicker within APPION to generate templates for subsequent RELION autopicking.
CTF correction
Software - Name: CTFFIND (ver. 4) / Software - details: run from within RELION Details: Correction within RELION based on values determined in CTFFIND4.
Details: Box and pixel size were adjusted before initial refinement.
Final reconstruction
Number classes used: 1 / Applied symmetry - Point group: C1 (asymmetric) / Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 5.4 Å / Resolution method: FSC 0.143 CUT-OFF / Software - Name: RELION (ver. 1.4) Details: RELION 3D auto-refinement (gold standard). The final 3D classification used signal-subtracted particle images. The final reconstruction used the corresponding un-subtracted particle images. Number images used: 35900
Initial angle assignment
Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) Details: Maximum-likelihood 3D classification within RELION.
Final angle assignment
Type: PROJECTION MATCHING / Software - Name: RELION (ver. 1.4) / Details: Maximum-likelihood 3D refinement within RELION.
Final 3D classification
Number classes: 6 / Software - Name: RELION (ver. 1.4) Details: Signal-subtracted classification in RELION 1.4, using 6 classes.
-
Atomic model buiding 1
Details
Fitting of a refined coordinate model obtained from a 4.4 Angstrom reconstruction of TFIIH from the same initial dataset (PDB ID 5OF4).
Refinement
Space: REAL / Protocol: RIGID BODY FIT
+
About Yorodumi
-
News
-
Feb 9, 2022. New format data for meta-information of EMDB entries
New format data for meta-information of EMDB entries
Version 3 of the EMDB header file is now the official format.
The previous official version 1.9 will be removed from the archive.
In the structure databanks used in Yorodumi, some data are registered as the other names, "COVID-19 virus" and "2019-nCoV". Here are the details of the virus and the list of structure data.
Jan 31, 2019. EMDB accession codes are about to change! (news from PDBe EMDB page)
EMDB accession codes are about to change! (news from PDBe EMDB page)
The allocation of 4 digits for EMDB accession codes will soon come to an end. Whilst these codes will remain in use, new EMDB accession codes will include an additional digit and will expand incrementally as the available range of codes is exhausted. The current 4-digit format prefixed with “EMD-” (i.e. EMD-XXXX) will advance to a 5-digit format (i.e. EMD-XXXXX), and so on. It is currently estimated that the 4-digit codes will be depleted around Spring 2019, at which point the 5-digit format will come into force.
The EM Navigator/Yorodumi systems omit the EMD- prefix.
Related info.:Q: What is EMD? / ID/Accession-code notation in Yorodumi/EM Navigator
Yorodumi is a browser for structure data from EMDB, PDB, SASBDB, etc.
This page is also the successor to EM Navigator detail page, and also detail information page/front-end page for Omokage search.
The word "yorodu" (or yorozu) is an old Japanese word meaning "ten thousand". "mi" (miru) is to see.
Related info.:EMDB / PDB / SASBDB / Comparison of 3 databanks / Yorodumi Search / Aug 31, 2016. New EM Navigator & Yorodumi / Yorodumi Papers / Jmol/JSmol / Function and homology information / Changes in new EM Navigator and Yorodumi
 Movie
Movie Controller
Controller

 Yorodumi
Yorodumi Open data
Open data
 Basic information
Basic information Map data
Map data Sample
Sample Function and homology information
Function and homology information Homo sapiens (human)
Homo sapiens (human) Authors
Authors United States, 1 items
United States, 1 items  Citation
Citation Structure visualization
Structure visualization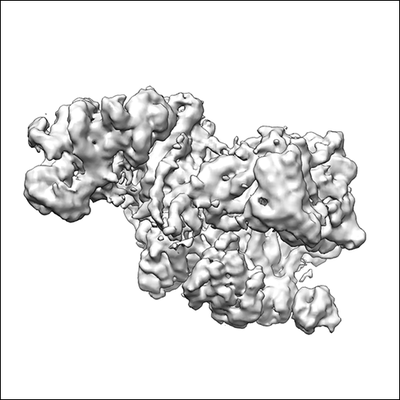
 Downloads & links
Downloads & links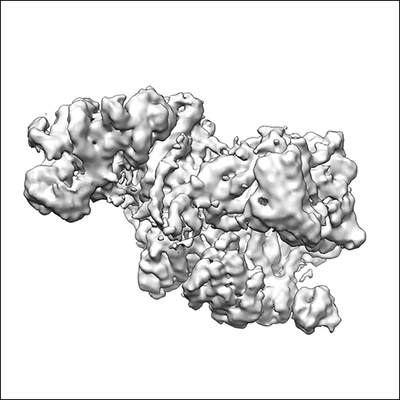 emd_8816.png
emd_8816.png http://ftp.pdbj.org/pub/emdb/structures/EMD-8816
http://ftp.pdbj.org/pub/emdb/structures/EMD-8816

























 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)



















 Sample components
Sample components Processing
Processing Electron microscopy
Electron microscopy FIELD EMISSION GUN
FIELD EMISSION GUN
