 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
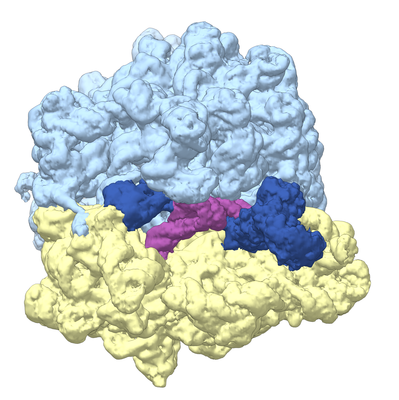
Yorodumi- EMDB-8108: Structure of RelA bound to the 70S ribosome (overall map, class 2) -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-8108 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of RelA bound to the 70S ribosome (overall map, class 2) | |||||||||
 Map data Map data | None | |||||||||
 Sample Sample |
| |||||||||
| Biological species |  | |||||||||
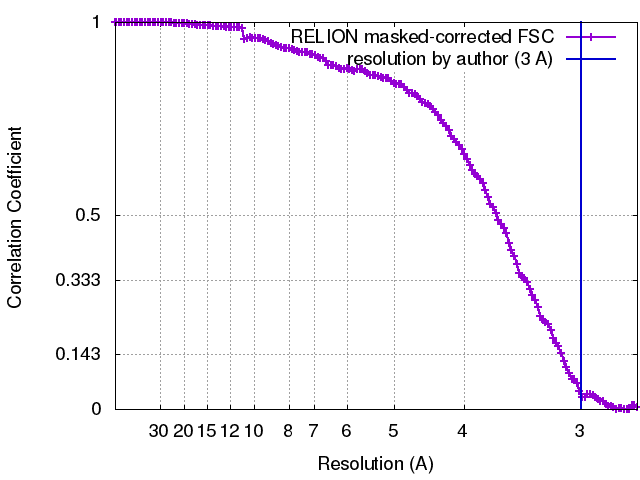
| Method | single particle reconstruction / cryo EM / Resolution: 3.0 Å | |||||||||
 Authors Authors | Brown A / Fernandez IS / Gordiyenko Y / Ramakrishnan V | |||||||||
 Citation Citation | Journal: Nature / Year: 2016 Title: Ribosome-dependent activation of stringent control. Authors: Alan Brown / Israel S Fernández / Yuliya Gordiyenko / V Ramakrishnan /  Abstract: In order to survive, bacteria continually sense, and respond to, environmental fluctuations. Stringent control represents a key bacterial stress response to nutrient starvation that leads to rapid ...In order to survive, bacteria continually sense, and respond to, environmental fluctuations. Stringent control represents a key bacterial stress response to nutrient starvation that leads to rapid and comprehensive reprogramming of metabolic and transcriptional patterns. In general, transcription of genes for growth and proliferation is downregulated, while those important for survival and virulence are upregulated. Amino acid starvation is sensed by depletion of the aminoacylated tRNA pools, and this results in accumulation of ribosomes stalled with non-aminoacylated (uncharged) tRNA in the ribosomal A site. RelA is recruited to stalled ribosomes and activated to synthesize a hyperphosphorylated guanosine analogue, (p)ppGpp, which acts as a pleiotropic secondary messenger. However, structural information about how RelA recognizes stalled ribosomes and discriminates against aminoacylated tRNAs is missing. Here we present the cryo-electron microscopy structure of RelA bound to the bacterial ribosome stalled with uncharged tRNA. The structure reveals that RelA utilizes a distinct binding site compared to the translational factors, with a multi-domain architecture that wraps around a highly distorted A-site tRNA. The TGS (ThrRS, GTPase and SpoT) domain of RelA binds the CCA tail to orient the free 3' hydroxyl group of the terminal adenosine towards a β-strand, such that an aminoacylated tRNA at this position would be sterically precluded. The structure supports a model in which association of RelA with the ribosome suppresses auto-inhibition to activate synthesis of (p)ppGpp and initiate the stringent response. Since stringent control is responsible for the survival of pathogenic bacteria under stress conditions, and contributes to chronic infections and antibiotic tolerance, RelA represents a good target for the development of novel antibacterial therapeutics. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
 Movie viewer Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_8108.map.gz | 8.7 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-8108-v30.xmlemd-8108.xml | 23.8 KB 23.8 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_8108_fsc.xml | 14.1 KB | Display | FSC data file |
| Images |  emd_8108.png emd_8108.png | 173.2 KB | ||
| Others | emd_8108_half_map_1.map.gzemd_8108_half_map_2.map.gz | 216.7 MB 216.7 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-8108ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8108 http://ftp.pdbj.org/pub/emdb/structures/EMD-8108ftp://ftp.pdbj.org/pub/emdb/structures/EMD-8108 | HTTPS FTP |
-Related structure data
| Related structure data |  8107C  8109C  8110C  8111C  8112C  8113C  8114C  8115C  5iqrC C: citing same article ( |
|---|---|
| Similar structure data |














-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |

-Map
| File | Download / File: emd_8108.map.gz / Format: CCP4 / Size: 244.1 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
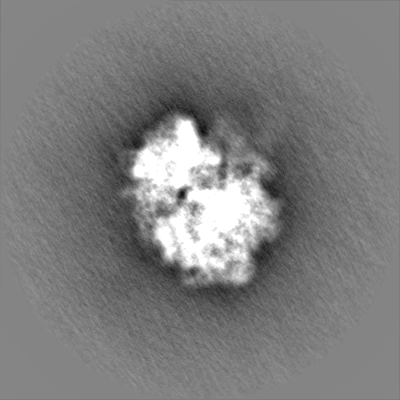
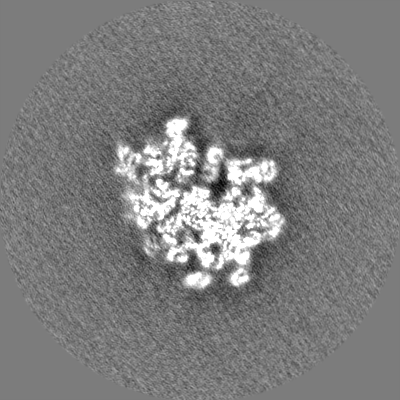
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.34 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
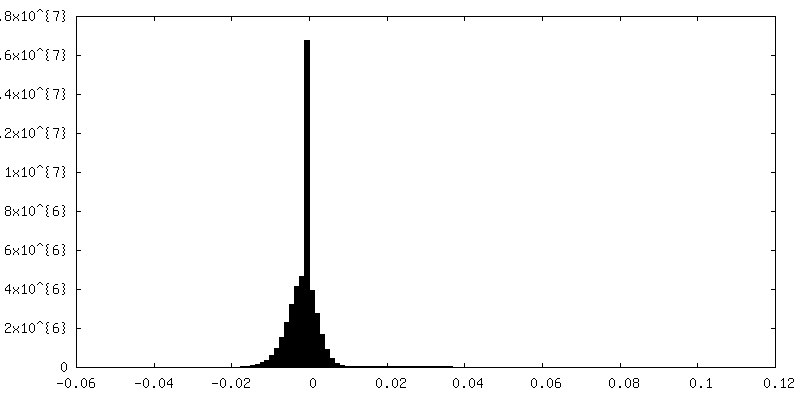
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)




















-Supplemental data
-Half map: None
| File | emd_8108_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||
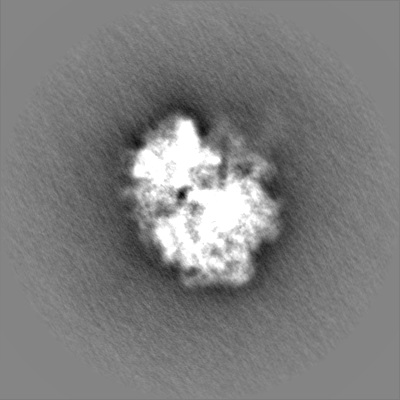
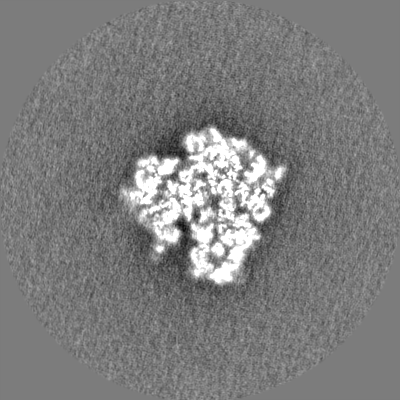
| Projections & Slices |
| ||||||||||||
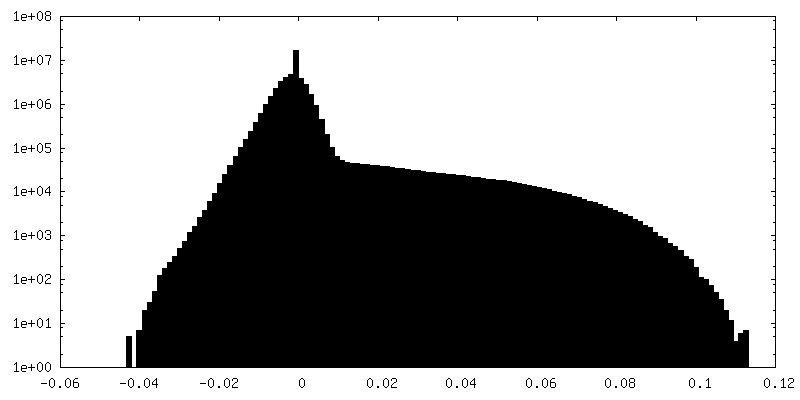
| Density Histograms |

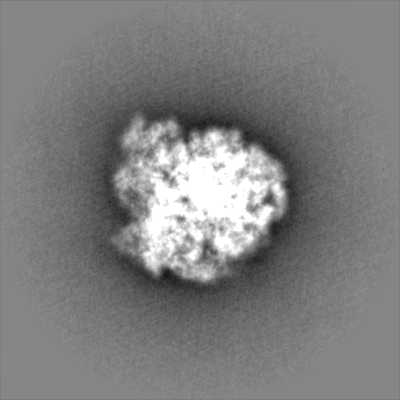
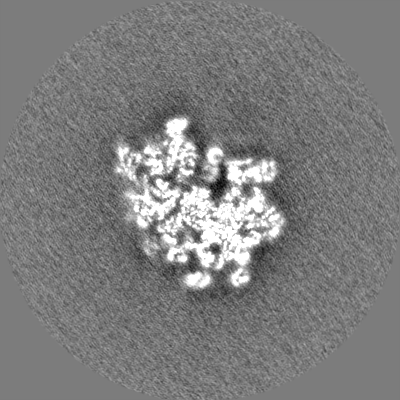
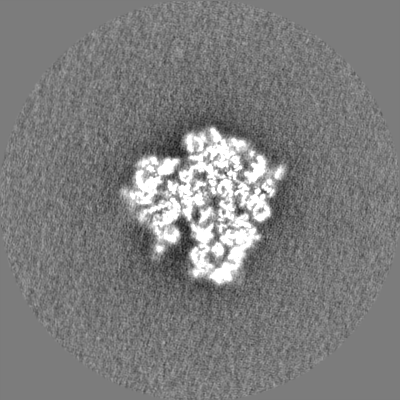
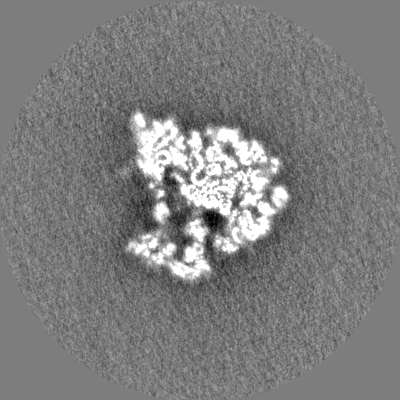
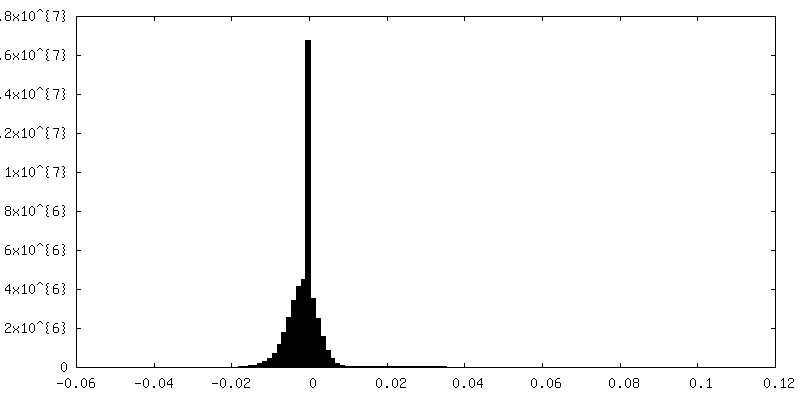
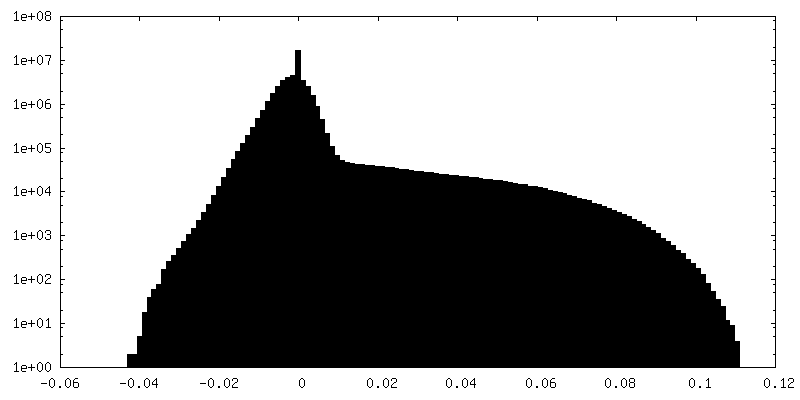
-Half map: None
| File | emd_8108_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | None | ||||||||||||
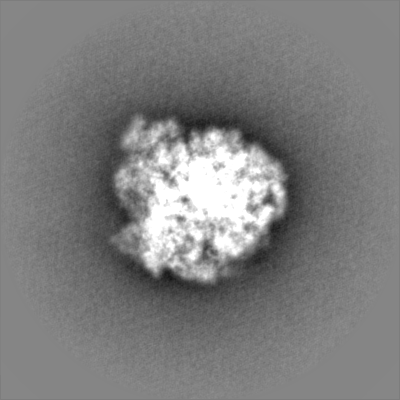
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : 70 S ribosome
| Entire | Name: 70 S ribosome |
|---|---|
| Components |
|
-Supramolecule #1: 70 S ribosome
| Supramolecule | Name: 70 S ribosome / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1-#59 |
|---|---|
| Source (natural) | Organism: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.1 mg/mL | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Buffer | pH: 7.5 Component:
| ||||||||||||||||||
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 400 / Support film - Material: CARBON / Support film - topology: CONTINUOUS / Support film - Film thickness: 3.0 nm / Pretreatment - Type: GLOW DISCHARGE | ||||||||||||||||||
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 100 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK II / Details: Grids were blotted for 5 s. | ||||||||||||||||||
| Details | This sample was monodisperse. |
- Electron microscopy
Electron microscopy
| Microscope | FEI POLARA 300 |
|---|---|
| Image recording | Film or detector model: OTHER / Average electron dose: 35.0 e/Å2 Details: FEI Falcon III Images were collected in movie-mode at 30 frames per second |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | Calibrated magnification: 104478 / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Nominal defocus max: 3.0 µm / Nominal defocus min: 1.8 µm |
| Sample stage | Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Tecnai Polara / Image courtesy: FEI Company |
+Image processing
-Atomic model buiding 1
| Initial model | PDB ID: |
|---|---|
| Refinement | Space: RECIPROCAL / Protocol: OTHER / Overall B value: 112.7 / Target criteria: Average FSC |

