 ムービー
ムービー コントローラー
コントローラー


+ データを開く
データを開く

- 基本情報
基本情報
| 登録情報 | データベース: EMDB / ID: EMD-5626 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| タイトル | Molecular Architecture of the ATP-Dependent Chromatin Remodeling Complex SWR1 by 3 Dimensional Electron Microscopy | |||||||||
 マップデータ マップデータ | 3D Cryo-negative EM structure of SWR1 | |||||||||
 試料 試料 |
| |||||||||
 キーワード キーワード | SWR1 / ATP-dependent chromatin remodeling / nucleosome / H2A.Z / Rvb1 / Rvb2 / AAA+ ATPases / 3 dimensional electron microscopy | |||||||||
| 生物種 |  | |||||||||
| 手法 | 単粒子再構成法 / クライオ電子顕微鏡法 / ネガティブ染色法 / 解像度: 28.0 Å | |||||||||
 データ登録者 データ登録者 | Nguyen VQ / Ranjan A / Stengel F / Wei D / Aebersold R / Wu C / Leschziner AE | |||||||||
 引用 引用 | ジャーナル: Cell / 年: 2013 タイトル: Molecular architecture of the ATP-dependent chromatin-remodeling complex SWR1. 著者: Vu Q Nguyen / Anand Ranjan / Florian Stengel / Debbie Wei / Ruedi Aebersold / Carl Wu / Andres E Leschziner /  要旨: The ATP-dependent chromatin-remodeling complex SWR1 exchanges a variant histone H2A.Z/H2B dimer for a canonical H2A/H2B dimer at nucleosomes flanking histone-depleted regions, such as promoters. This ...The ATP-dependent chromatin-remodeling complex SWR1 exchanges a variant histone H2A.Z/H2B dimer for a canonical H2A/H2B dimer at nucleosomes flanking histone-depleted regions, such as promoters. This localization of H2A.Z is conserved throughout eukaryotes. SWR1 is a 1 megadalton complex containing 14 different polypeptides, including the AAA+ ATPases Rvb1 and Rvb2. Using electron microscopy, we obtained the three-dimensional structure of SWR1 and mapped its major functional components. Our data show that SWR1 contains a single heterohexameric Rvb1/Rvb2 ring that, together with the catalytic subunit Swr1, brackets two independently assembled multisubunit modules. We also show that SWR1 undergoes a large conformational change upon engaging a limited region of the nucleosome core particle. Our work suggests an important structural role for the Rvbs and a distinct substrate-handling mode by SWR1, thereby providing a structural framework for understanding the complex dimer-exchange reaction. | |||||||||
| 履歴 |
|
- 構造の表示
構造の表示
| ムービー |
 ムービービューア ムービービューア |
|---|---|
| 構造ビューア | EMマップ: SurfViewMolmilJmol/JSmol |
| 添付画像 |

- ダウンロードとリンク
ダウンロードとリンク
-EMDBアーカイブ
| マップデータ | emd_5626.map.gz | 12 MB | EMDBマップデータ形式 | |
|---|---|---|---|---|
| ヘッダ (付随情報) | emd-5626-v30.xmlemd-5626.xml | 12.4 KB 12.4 KB | 表示 表示 | EMDBヘッダ |
| 画像 |  emd_5626.png emd_5626.png | 79.4 KB | ||
| アーカイブディレクトリ |  http://ftp.pdbj.org/pub/emdb/structures/EMD-5626ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5626 http://ftp.pdbj.org/pub/emdb/structures/EMD-5626ftp://ftp.pdbj.org/pub/emdb/structures/EMD-5626 | HTTPS FTP |
-関連構造データ









-リンク
| EMDBのページ | EMDB (EBI/PDBe) / EMDataResource |
|---|
-マップ
| ファイル | ダウンロード / ファイル: emd_5626.map.gz / 形式: CCP4 / 大きさ: 12.6 MB / タイプ: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
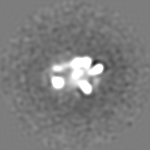
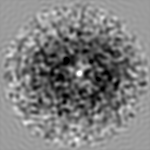
| 注釈 | 3D Cryo-negative EM structure of SWR1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 投影像・断面図 | 画像のコントロール
画像は Spider により作成 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ボクセルのサイズ | X=Y=Z: 3.45 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
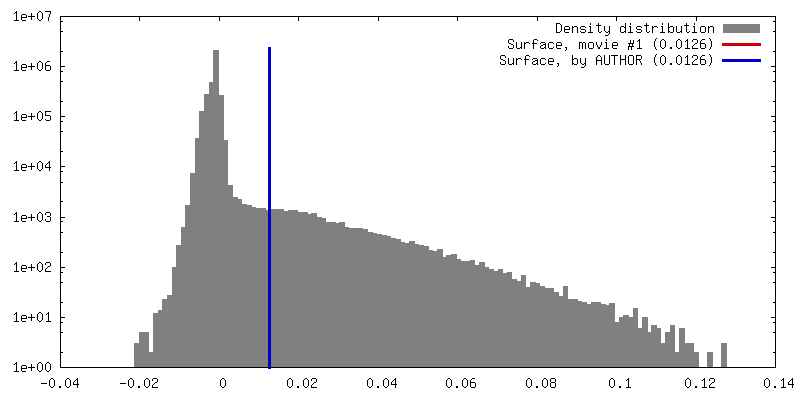
| 密度 |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 対称性 | 空間群: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 詳細 | EMDB XML:
CCP4マップ ヘッダ情報:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)


















-添付データ
- 試料の構成要素
試料の構成要素
-全体 : ATP-dependent chromatin remodeling complex SWR1
| 全体 | 名称: ATP-dependent chromatin remodeling complex SWR1 |
|---|---|
| 要素 |
|
-超分子 #1000: ATP-dependent chromatin remodeling complex SWR1
| 超分子 | 名称: ATP-dependent chromatin remodeling complex SWR1 / タイプ: sample / ID: 1000 / Number unique components: 14 |
|---|---|
| 分子量 | 理論値: 1.0 MDa |
-分子 #1: SWR1
| 分子 | 名称: SWR1 / タイプ: protein_or_peptide / ID: 1 / 組換発現: No / データベース: NCBI |
|---|---|
| 由来(天然) | 生物種: |
| 分子量 | 理論値: 1.0 MDa |
-実験情報
-構造解析
| 手法 | ネガティブ染色法, クライオ電子顕微鏡法 |
|---|---|
 解析 解析 | 単粒子再構成法 |
| 試料の集合状態 | particle |
-試料調製
| 緩衝液 | pH: 7.6 詳細: 25 mM HEPES-KOH, 1 mM EDTA, 2 mM MgCl2, 0.01% NP-40, 1 mM DTT, 100 mM KCl |
|---|---|
| 染色 | タイプ: NEGATIVE 詳細: Sample was adsorbed for 15-30 minutes at 4 degrees C. Grid was rinsed with drops of stain (2% uranyl formate) and then a second layer of thin carbon was floated onto the grid. After blotting, ...詳細: Sample was adsorbed for 15-30 minutes at 4 degrees C. Grid was rinsed with drops of stain (2% uranyl formate) and then a second layer of thin carbon was floated onto the grid. After blotting, the grid was frozen in liquid nitrogen. |
| グリッド | 詳細: Cu 200-mesh Quantifoil grids with thin carbon support, glow discharged in air |
| 凍結 | 凍結剤: NITROGEN / 装置: OTHER / 手法: Cryo-negative staining with manual freezing |
- 電子顕微鏡法
電子顕微鏡法
| 顕微鏡 | FEI TECNAI F20 |
|---|---|
| 温度 | 平均: 100 K |
| 日付 | 2012年5月20日 |
| 撮影 | デジタル化 - サンプリング間隔: 15 µm / 実像数: 300 / 平均電子線量: 20 e/Å2 / カメラ長: 15 |
| 電子線 | 加速電圧: 120 kV / 電子線源:  FIELD EMISSION GUN FIELD EMISSION GUN |
| 電子光学系 | 倍率(補正後): 87000 / 照射モード: FLOOD BEAM / 撮影モード: BRIGHT FIELD / Cs: 2.0 mm / 最大 デフォーカス(公称値): 2.0 µm / 最小 デフォーカス(公称値): 0.5 µm / 倍率(公称値): 62000 |
| 試料ステージ | 試料ホルダーモデル: GATAN LIQUID NITROGEN |
| 実験機器 |  モデル: Tecnai F20 / 画像提供: FEI Company |
-画像解析
| 詳細 | In order to extract the molecular images from the micrographs, particles were windowed out in one set of micrographs (-45 degrees) using the Boxer interface in EMAN1. Custom-built SPIDER scripts were used to calculate alignment parameters between the +45 degree and -45 degree micrographs and to extract the tilt mates in the +45 degree micrographs. The Contrast Transfer Function (CTF) was estimated and corrected for using the program CTFTILT and the SPIDER command TF CT. Single particles were binned by 2, resulting in a pixel size of 3.3 Angstrom. The +45 degree and -45 degree datasets were combined into a stack of ~32,000 particles and 2D alignment and classification were performed in IMAGIC. Initial models were computed from classes containing 100-200 members using the Orthogonal Tilt Reconstruction approach as described. For projection-matching refinement, the OTR models were initially refined against 2D class averages of cryo-negative data. To generate the class averages, particles were extracted from the micrographs as described above and CTF estimation and phase flipping were performed using the EMAN2 workflow. The particles were then binned by 2, resulting in a pixel size of 3.45 Angstrom. Approximately 32,000 particles were subjected to reference-free 2D alignment and classification in IMAGIC. In order to minimize heterogeneity, classes were generated with relatively few (15-20) particles. The OTR models were filtered to 80 Angstrom resolution and 15-23 iterations of projection matching refinement were performed using angular steps of 250, 200, 150, 100, and 80-50 against 2D class averages in SPIDER using the AP SH and BP 32F commands. To minimize noise in the reconstructions, a threshold mask calculated for 500% to 150%of the theoretical molecular weight of the sample (1.0 MDa) was applied. The mask was gradually tightened throughout refinement and its filtration was determined by the resolution of the 3D map, computed according to the 0.5 FSC criterion. Refinement results were stable after 15 iterations, and the resolutions of the 3D maps were 50-60 Angstrom. The resulting 3D maps (without additional filtration) were then similarly refined against single cryo-negative particles. For this step, 15 iterations of projection-matching refinement were carried out at angular steps of 250, 210, 180, 150, 130, 110 and 100-40. Threshold masks computed for 500% to 100% of the MW were also utilized. |
|---|---|
| CTF補正 | 詳細: CTFTILT (Grigorieff) for initial model; EMAN2 for projection-matching refinement |
| 最終 再構成 | アルゴリズム: OTHER / 解像度のタイプ: BY AUTHOR / 解像度: 28.0 Å / 解像度の算出法: FSC 0.5 CUT-OFF / ソフトウェア - 名称: EMAN1, EMAN2, IMAGIC, SPIDER / 使用した粒子像数: 18000 |
| 最終 角度割当 | 詳細: SPIDER |
