 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-46770: Human Adenovirus 657 (Ad657) and Coagulation factor II (Prothromb... -
+ Open data
Open data

- Basic information
Basic information
| Entry |  | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Human Adenovirus 657 (Ad657) and Coagulation factor II (Prothrombin) at 4.17A resolution. | |||||||||
 Map data Map data | SCAd657_II_virion_4.17A | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Human adenovirus (Ad) 657 Blood Coagulation Factor II Binding Complex / VIRUS | |||||||||
| Biological species |  Human adenovirus 57 Human adenovirus 57 | |||||||||
| Method | single particle reconstruction / cryo EM / Resolution: 4.17 Å | |||||||||
 Authors Authors | Reddy VS / Ma OX | |||||||||
| Funding support |  United States, 1 items United States, 1 items
| |||||||||
 Citation Citation | Journal: Nat Commun / Year: 2024 Title: Structure-derived insights from blood factors binding to the surfaces of different adenoviruses. Authors: Haley E Mudrick / Shao-Chia Lu / Janarjan Bhandari / Mary E Barry / Jack R Hemsath / Felix G M Andres / Olivia X Ma / Michael A Barry / Vijay S Reddy / Abstract: The tropism of adenoviruses (Ads) is significantly influenced by the binding of various blood factors. To investigate differences in their binding, we conducted cryo-EM analysis on complexes of ...The tropism of adenoviruses (Ads) is significantly influenced by the binding of various blood factors. To investigate differences in their binding, we conducted cryo-EM analysis on complexes of several human adenoviruses with human platelet factor-4 (PF4), coagulation factors FII (Prothrombin), and FX. While we observed EM densities for FII and FX bound to all the species-C adenoviruses examined, no densities were seen for PF4, even though PF4 can co-pellet with various Ads. Similar to FX, the γ-carboxyglutamic acid (Gla) domain of FII binds within the surface cavity of hexon trimers. While FII binds equally to species-C Ads: Ad5, Ad6, and Ad657, FX exhibits significantly better binding to Ad5 and Ad657 compared to Ad6. Although only the FX-Gla domain is observed at high-resolution (3.7 Å), the entire FX is visible at low-resolution bound to Ad5 in three equivalent binding modes consistent with the 3-fold symmetric hexon. Only the Gla and kringle-1 domains of FII are visible on all the species-C adenoviruses, where the rigid FII binds in an upright fashion, in contrast to the flexible and bent FX. These data suggest that differential binding of FII and FX may shield certain species-C adenoviruses differently against immune molecules, thereby modulating their tropism. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Supplemental images |
|---|

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_46770.map.gz | 880 MB |  EMDB map data format EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-46770-v30.xmlemd-46770.xml | 14.4 KB 14.4 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_46770_fsc.xml | 28.2 KB | Display | FSC data file |
| Images |  emd_46770.png emd_46770.png | 173.7 KB | ||
| Filedesc metadata | emd-46770.cif.gz | 4.5 KB | ||
| Others | emd_46770_half_map_1.map.gzemd_46770_half_map_2.map.gz | 1.5 GB 1.5 GB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-46770ftp://ftp.pdbj.org/pub/emdb/structures/EMD-46770 http://ftp.pdbj.org/pub/emdb/structures/EMD-46770ftp://ftp.pdbj.org/pub/emdb/structures/EMD-46770 | HTTPS FTP |
-Related structure data
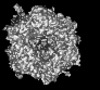
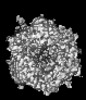
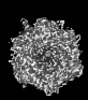
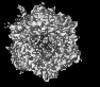
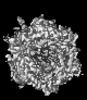











-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|
-Map
| File | Download / File: emd_46770.map.gz / Format: CCP4 / Size: 976.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | SCAd657_II_virion_4.17A | ||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 1.408 Å | ||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
|
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Half map: SCAd657 II virion 4.17A
| File | emd_46770_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | SCAd657_II_virion_4.17A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








-Half map: SCAd657 II virion 4.17A
| File | emd_46770_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | SCAd657_II_virion_4.17A | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |








- Sample components
Sample components
-Entire : Human adenovirus 657 (Ad657) in complex with Coagulation factor F...
| Entire | Name: Human adenovirus 657 (Ad657) in complex with Coagulation factor FII (Prothrombin) |
|---|---|
| Components |
|
-Supramolecule #1: Human adenovirus 657 (Ad657) in complex with Coagulation factor F...
| Supramolecule | Name: Human adenovirus 657 (Ad657) in complex with Coagulation factor FII (Prothrombin) type: complex / ID: 1 / Parent: 0 / Details: Icosahedral (I1) reconstruction |
|---|---|
| Source (natural) | Organism: Human adenovirus 57 |
| Molecular weight | Theoretical: 150 MDa |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 2 mg/mL |
|---|---|
| Buffer | pH: 7.2 Details: 50mM HEPES pH 7.2, 300mM NaCl, 10mM CaCl2 and 5mM MgCl2 |
| Grid | Model: Quantifoil R1.2/1.3 / Support film - Material: CARBON |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 90 % / Chamber temperature: 277 K |
| Details | Human Adenovirus 657 in complex with coagulation Factor X |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Image recording | Film or detector model: GATAN K3 BIOCONTINUUM (6k x 4k) / Number grids imaged: 2 / Number real images: 7281 / Average exposure time: 5.0 sec. / Average electron dose: 80.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 3.0 µm / Nominal defocus min: 0.8 µm |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |
+Image processing

-Atomic model buiding 1
| Refinement | Protocol: RIGID BODY FIT / Target criteria: Cross-correlation Coefficient |
|---|
