 Movie
Movie Controller
Controller

[English] 日本語
 Yorodumi
Yorodumi- EMDB-22463: Proteinase K soaked with I3C determined by MicroED from a single ... -
+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-22463 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Proteinase K soaked with I3C determined by MicroED from a single milled microcrystal | |||||||||
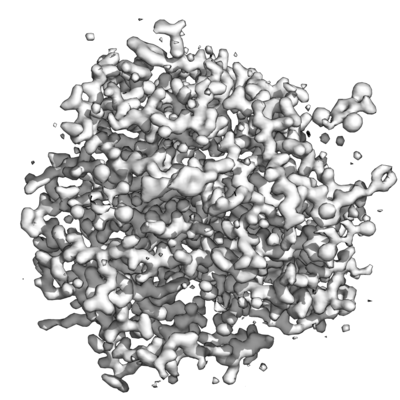
 Map data Map data | 2Fo-Fc map | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | Proteinase K / HYDROLASE | |||||||||
| Function / homology |  Function and homology information Function and homology informationpeptidase K / serine-type endopeptidase activity / proteolysis / extracellular region / metal ion binding Similarity search - Function | |||||||||
| Biological species |  Parengyodontium album (fungus) Parengyodontium album (fungus) | |||||||||
| Method | electron crystallography / cryo EM / Resolution: 1.78 Å | |||||||||
 Authors Authors | Martynowycz MW / Gonen T | |||||||||
| Funding support |  United States, 2 items United States, 2 items
| |||||||||
 Citation Citation | Journal: Structure / Year: 2021 Title: Ligand Incorporation into Protein Microcrystals for MicroED by On-Grid Soaking. Authors: Michael W Martynowycz / Tamir Gonen / Abstract: A high throughout method for soaking ligands into protein microcrystals on TEM grids is presented. Every crystal on the grid is soaked simultaneously using only standard cryoelectron microscopy ...A high throughout method for soaking ligands into protein microcrystals on TEM grids is presented. Every crystal on the grid is soaked simultaneously using only standard cryoelectron microscopy vitrification equipment. The method is demonstrated using proteinase K microcrystals soaked with the 5-amino-2,4,6-triodoisophthalic acid (I3C) magic triangle. A soaked microcrystal is milled to a thickness of approximately 200 nm using a focused ion beam, and MicroED data are collected. A high-resolution structure of the protein with four ligands at high occupancy is determined. Both the number of ligands bound and their occupancy is higher using on-grid soaking of microcrystals compared with much larger crystals treated similarly and investigated by X-ray crystallography. These results indicate that on-grid soaking ligands into microcrystals results in efficient uptake of ligands into protein microcrystals. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |
- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_22463.map.gz | 6 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-22463-v30.xmlemd-22463.xml | 13.6 KB 13.6 KB | Display Display | EMDB header |
| Images |  emd_22463.png emd_22463.png | 140.9 KB | ||
| Filedesc metadata | emd-22463.cif.gz | 5.5 KB | ||
| Filedesc structureFactors | emd_22463_sf.cif.gz | 553.8 KB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22463ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22463 http://ftp.pdbj.org/pub/emdb/structures/EMD-22463ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22463 | HTTPS FTP |
-Related structure data
| Related structure data |  7jsyMC M: atomic model generated by this map C: citing same article ( |
|---|---|
| Similar structure data |








-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |


-Map
| File | Download / File: emd_22463.map.gz / Format: CCP4 / Size: 6.6 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | 2Fo-Fc map | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. generated in cubic-lattice coordinate | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.56296 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 X (Sec.)
X (Sec.) Y (Row.)
Y (Row.) Z (Col.)
Z (Col.)





















-Supplemental data
- Sample components
Sample components
-Entire : Proteinase K
| Entire | Name: Proteinase K |
|---|---|
| Components |
|
-Supramolecule #1: Proteinase K
| Supramolecule | Name: Proteinase K / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 / Details: Serine protease |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28.9 KDa |
-Macromolecule #1: Proteinase K
| Macromolecule | Name: Proteinase K / type: protein_or_peptide / ID: 1 / Number of copies: 1 / Enantiomer: LEVO / EC number: peptidase K |
|---|---|
| Source (natural) | Organism: Parengyodontium album (fungus) |
| Molecular weight | Theoretical: 28.930783 KDa |
| Sequence | String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN ...String: AAQTNAPWGL ARISSTSPGT STYYYDESAG QGSCVYVIDT GIEASHPEFE GRAQMVKTYY YSSRDGNGHG THCAGTVGSR TYGVAKKTQ LFGVKVLDDN GSGQYSTIIA GMDFVASDKN NRNCPKGVVA SLSLGGGYSS SVNSAAARLQ SSGVMVAVAA G NNNADARN YSPASEPSVC TVGASDRYDR RSSFSNYGSV LDIFGPGTSI LSTWIGGSTR SISGTSMATP HVAGLAAYLM TL GKTTAAS ACRYIADTAN KGDLSNIPFG TVNLLAYNNY QA UniProtKB: Proteinase K |
-Macromolecule #2: 5-amino-2,4,6-triiodobenzene-1,3-dicarboxylic acid
| Macromolecule | Name: 5-amino-2,4,6-triiodobenzene-1,3-dicarboxylic acid / type: ligand / ID: 2 / Number of copies: 4 / Formula: I3C |
|---|---|
| Molecular weight | Theoretical: 558.835 Da |
| Chemical component information | 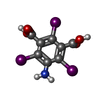 ChemComp-I3C: |
-Macromolecule #3: water
| Macromolecule | Name: water / type: ligand / ID: 3 / Number of copies: 246 / Formula: HOH |
|---|---|
| Molecular weight | Theoretical: 18.015 Da |
| Chemical component information |  ChemComp-HOH: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | electron crystallography |
| Aggregation state | 3D array |
-Sample preparation
| Concentration | 5 mg/mL |
|---|---|
| Buffer | pH: 7.5 |
| Grid | Model: Quantifoil R2/2 / Material: COPPER / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. |
| Vitrification | Cryogen name: ETHANE |
- Electron microscopy
Electron microscopy
| Microscope | TFS TALOS |
|---|---|
| Temperature | Min: 70.0 K |
| Details | Tilt series: -30 to +30, 0.25 deg/sec, 1 sec readout |
| Image recording | Film or detector model: FEI CETA (4k x 4k) / Digitization - Dimensions - Width: 4096 pixel / Digitization - Dimensions - Height: 4096 pixel / Number grids imaged: 1 / Number real images: 240 / Number diffraction images: 240 / Average exposure time: 1.0 sec. / Average electron dose: 0.01 e/Å2 |
| Electron beam | Acceleration voltage: 200 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 70.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: DIFFRACTION / Nominal defocus min: 0.0 µm / Camera length: 1900 mm |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN / Tilt angle: -30.0, 30.0 |
-Image processing
| Final reconstruction | Algorithm: FOURIER SPACE / Resolution.type: BY AUTHOR / Resolution: 1.78 Å / Resolution method: DIFFRACTION PATTERN/LAYERLINES |
|---|---|
| Crystallography statistics | Number intensities measured: 109326 / Number structure factors: 10458 / Fourier space coverage: 94.52 / R sym: 0.13 / R merge: 0.26 / Overall phase error: 18 / Overall phase residual: 18 / Phase error rejection criteria: 0 / High resolution: 1.78 Å / Shell - Shell ID: 1 / Shell - High resolution: 43.32 Å / Shell - Low resolution: 1.78 Å / Shell - Number structure factors: 10458 / Shell - Phase residual: 18 / Shell - Fourier space coverage: 94.52 / Shell - Multiplicity: 4.9 |
-Atomic model buiding 1
| Initial model | PDB ID: Chain - Chain ID: A / Chain - Residue range: 1-279 / Chain - Source name: PDB / Chain - Initial model type: experimental model |
|---|---|
| Refinement | Space: RECIPROCAL / Protocol: RIGID BODY FIT / Overall B value: 13.51 / Target criteria: maximum liklihood |
| Output model | PDB-7jsy: |

