Movie
Movie Controller
Controller


+ Open data
Open data

- Basic information
Basic information
| Entry | Database: EMDB / ID: EMD-22256 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Title | Structure of SARS-CoV-2 spike at pH 5.5, all RBDs down | |||||||||
 Map data Map data | Refined map after post-processing and masking | |||||||||
 Sample Sample |
| |||||||||
 Keywords Keywords | SARS-CoV-2 spike / COVID19 / VIRAL PROTEIN | |||||||||
| Function / homology |  Function and homology information Function and homology informationsymbiont-mediated disruption of host tissue / Maturation of spike protein / Translation of Structural Proteins / Virion Assembly and Release / host cell surface / Lectin pathway of complement activation / host extracellular region / symbiont-mediated-mediated suppression of host tetherin activity / Induction of Cell-Cell Fusion / structural constituent of virion ...symbiont-mediated disruption of host tissue / Maturation of spike protein / Translation of Structural Proteins / Virion Assembly and Release / host cell surface / Lectin pathway of complement activation / host extracellular region / symbiont-mediated-mediated suppression of host tetherin activity / Induction of Cell-Cell Fusion / structural constituent of virion / positive regulation of viral entry into host cell / Initial triggering of complement / membrane fusion / host cell endoplasmic reticulum-Golgi intermediate compartment membrane / Attachment and Entry / entry receptor-mediated virion attachment to host cell / receptor-mediated virion attachment to host cell / host cell surface receptor binding / symbiont-mediated suppression of host innate immune response / endocytosis involved in viral entry into host cell / receptor ligand activity / fusion of virus membrane with host plasma membrane / fusion of virus membrane with host endosome membrane / viral envelope / symbiont entry into host cell / virion attachment to host cell / host cell plasma membrane / SARS-CoV-2 activates/modulates innate and adaptive immune responses / virion membrane / membrane / identical protein binding / plasma membrane Similarity search - Function | |||||||||
| Biological species |   Severe acute respiratory syndrome coronavirus 2 Severe acute respiratory syndrome coronavirus 2 | |||||||||
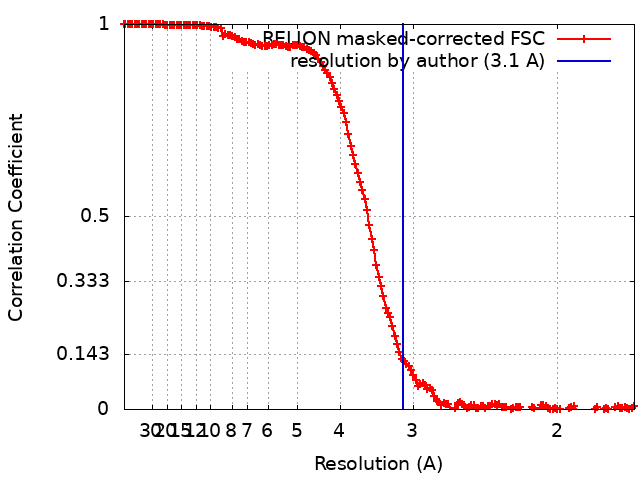
| Method | single particle reconstruction / cryo EM / Resolution: 3.1 Å | |||||||||
 Authors Authors | Tsybovsky Y / Zhou T / Olia A / Kwong PD | |||||||||
 Citation Citation | Journal: bioRxiv / Year: 2020 Title: Cryo-EM Structures Delineate a pH-Dependent Switch that Mediates Endosomal Positioning of SARS-CoV-2 Spike Receptor-Binding Domains. Abstract: The SARS-CoV-2 spike employs mobile receptor-binding domains (RBDs) to engage the ACE2 receptor and to facilitate virus entry. Antibodies can engage RBD but some, such as CR3022, fail to inhibit ...The SARS-CoV-2 spike employs mobile receptor-binding domains (RBDs) to engage the ACE2 receptor and to facilitate virus entry. Antibodies can engage RBD but some, such as CR3022, fail to inhibit entry despite nanomolar spike affinity. Here we show the SARS-CoV-2 spike to have low unfolding enthalpy at serological pH and up to 10-times more unfolding enthalpy at endosomal pH, where we observe significantly reduced CR3022 affinity. Cryo-EM structures -at serological and endosomal pH- delineated spike recognition of up to three ACE2 molecules, revealing RBD to freely adopt the 'up' conformation. In the absence of ACE2, single-RBD-up conformations dominated at pH 5.5, resolving into a locked all-down conformation at lower pH. Notably, a pH-dependent refolding region (residues 824-858) at the spike-interdomain interface displayed dramatic structural rearrangements and mediated RBD positioning and spike shedding of antibodies like CR3022. An endosomal mechanism involving spike-conformational change can thus facilitate immune evasion from RBD-'up'-recognizing antibody. | |||||||||
| History |
|
- Structure visualization
Structure visualization
| Movie |
Movie viewer |
|---|---|
| Structure viewer | EM map: SurfViewMolmilJmol/JSmol |
| Supplemental images |

- Downloads & links
Downloads & links
-EMDB archive
| Map data | emd_22256.map.gz | 20.2 MB | EMDB map data format | |
|---|---|---|---|---|
| Header (meta data) | emd-22256-v30.xmlemd-22256.xml | 25.9 KB 25.9 KB | Display Display | EMDB header |
| FSC (resolution estimation) | emd_22256_fsc.xml | 15.6 KB | Display | FSC data file |
| Images |  emd_22256.png emd_22256.png | 42.4 KB | ||
| Masks | emd_22256_msk_1.map | 325 MB | Mask map | |
| Filedesc metadata | emd-22256.cif.gz | 7.7 KB | ||
| Others | emd_22256_additional.map.gzemd_22256_half_map_1.map.gzemd_22256_half_map_2.map.gz | 260.5 MB 261.5 MB 261.5 MB | ||
| Archive directory |  http://ftp.pdbj.org/pub/emdb/structures/EMD-22256ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22256 http://ftp.pdbj.org/pub/emdb/structures/EMD-22256ftp://ftp.pdbj.org/pub/emdb/structures/EMD-22256 | HTTPS FTP |
-Related structure data
| Related structure data |  6xm5MC  6xluC  6xm0C  6xm3C  6xm4C  7jwyC  7kmbC  7kmsC  7kmzC  7knbC  7kneC  7knhC  7kniC C: citing same article ( M: atomic model generated by this map |
|---|---|
| Similar structure data |
















-Links
| EMDB pages | EMDB (EBI/PDBe) / EMDataResource |
|---|---|
| Related items in Molecule of the Month |






-Map
| File | Download / File: emd_22256.map.gz / Format: CCP4 / Size: 325 MB / Type: IMAGE STORED AS FLOATING POINT NUMBER (4 BYTES) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Refined map after post-processing and masking | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Projections & slices | Image control
Images are generated by Spider. | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Voxel size | X=Y=Z: 0.85 Å | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
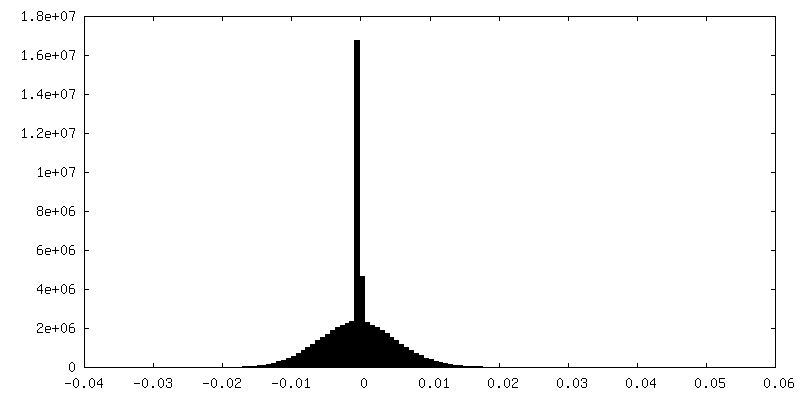
| Density |
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Symmetry | Space group: 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Details | EMDB XML:
CCP4 map header:
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 Z (Sec.)
Z (Sec.) Y (Row.)
Y (Row.) X (Col.)
X (Col.)





















-Supplemental data
-Mask #1
| File | emd_22256_msk_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Projections & Slices |
| ||||||||||||
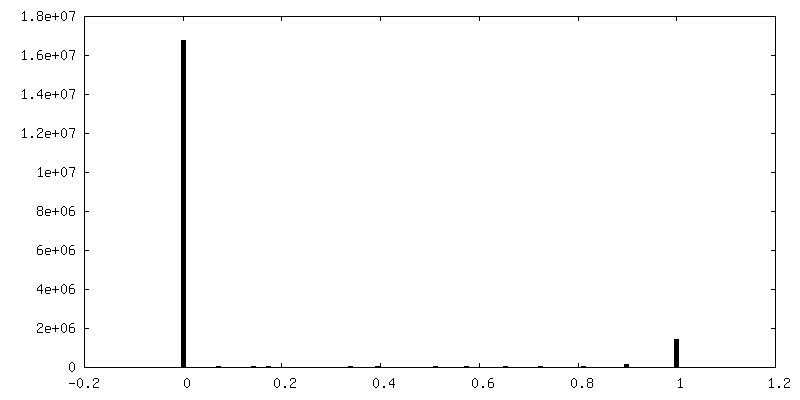
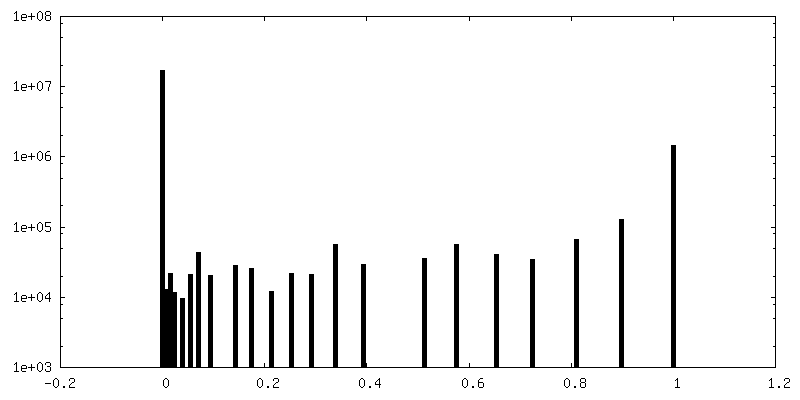
| Density Histograms |






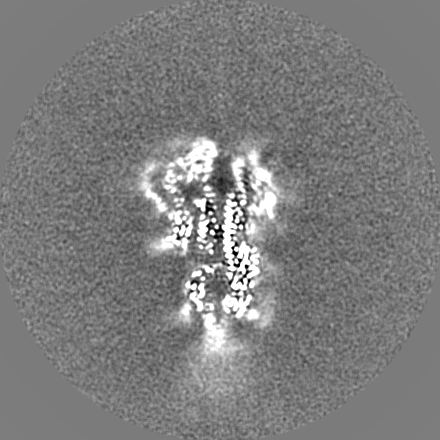
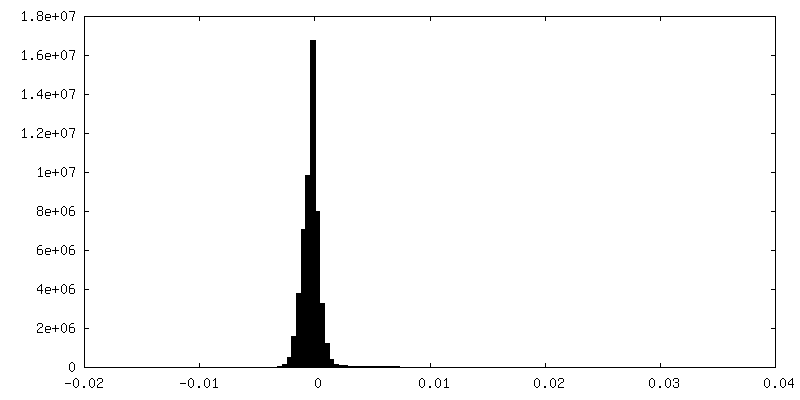
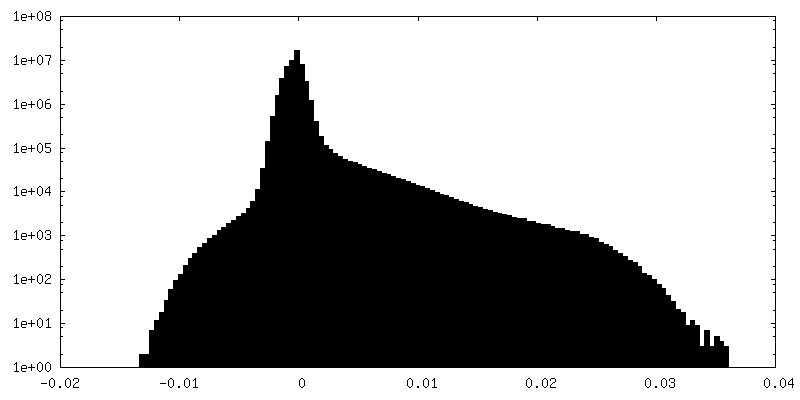
-Additional map: Refined map before post-processing
| File | emd_22256_additional.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Refined map before post-processing | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




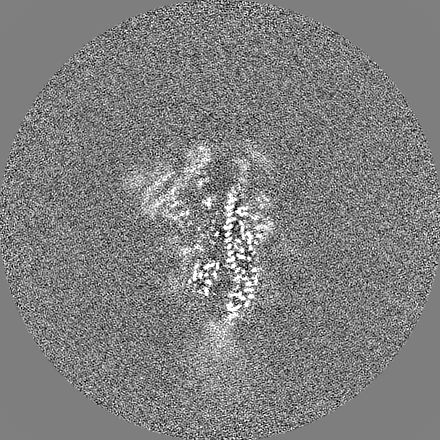
-Half map: Half-map 1
| File | emd_22256_half_map_1.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map 1 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |




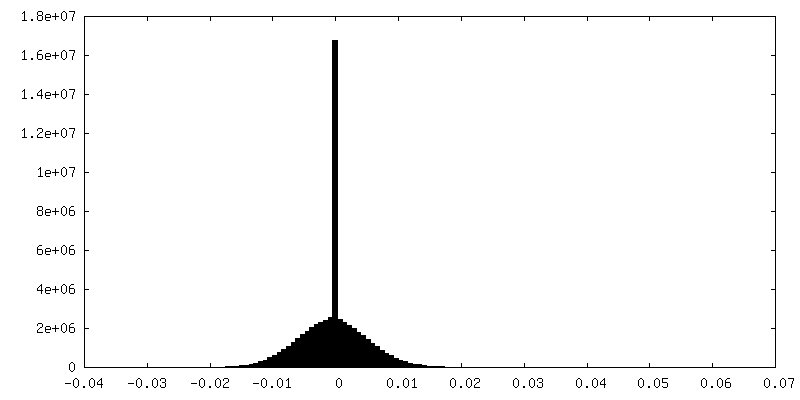
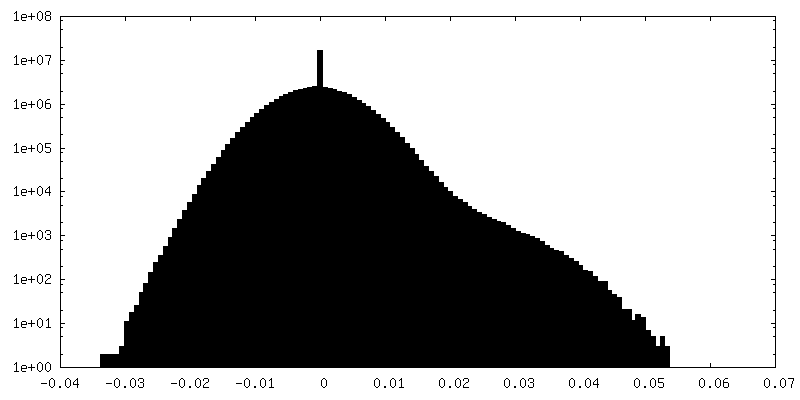
-Half map: Half-map 2
| File | emd_22256_half_map_2.map | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Annotation | Half-map 2 | ||||||||||||
| Projections & Slices |
| ||||||||||||
| Density Histograms |


- Sample components
Sample components
-Entire : SARS-CoV-2 Spike
| Entire | Name: SARS-CoV-2 Spike |
|---|---|
| Components |
|
-Supramolecule #1: SARS-CoV-2 Spike
| Supramolecule | Name: SARS-CoV-2 Spike / type: complex / ID: 1 / Parent: 0 / Macromolecule list: #1 |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome coronavirus 2 |
| Molecular weight | Theoretical: 423 KDa |
-Macromolecule #1: Spike glycoprotein
| Macromolecule | Name: Spike glycoprotein / type: protein_or_peptide / ID: 1 / Number of copies: 3 / Enantiomer: LEVO |
|---|---|
| Source (natural) | Organism: Severe acute respiratory syndrome coronavirus 2 |
| Molecular weight | Theoretical: 140.952531 KDa |
| Recombinant expression | Organism:  Homo sapiens (human) Homo sapiens (human) |
| Sequence | String: QCVNLTTRTQ LPPAYTNSFT RGVYYPDKVF RSSVLHSTQD LFLPFFSNVT WFHAIHVSGT NGTKRFDNPV LPFNDGVYFA STEKSNIIR GWIFGTTLDS KTQSLLIVNN ATNVVIKVCE FQFCNDPFLG VYYHKNNKSW MESEFRVYSS ANNCTFEYVS Q PFLMDLEG ...String: QCVNLTTRTQ LPPAYTNSFT RGVYYPDKVF RSSVLHSTQD LFLPFFSNVT WFHAIHVSGT NGTKRFDNPV LPFNDGVYFA STEKSNIIR GWIFGTTLDS KTQSLLIVNN ATNVVIKVCE FQFCNDPFLG VYYHKNNKSW MESEFRVYSS ANNCTFEYVS Q PFLMDLEG KQGNFKNLRE FVFKNIDGYF KIYSKHTPIN LVRDLPQGFS ALEPLVDLPI GINITRFQTL LALHRSYLTP GD SSSGWTA GAAAYYVGYL QPRTFLLKYN ENGTITDAVD CALDPLSETK CTLKSFTVEK GIYQTSNFRV QPTESIVRFP NIT NLCPFG EVFNATRFAS VYAWNRKRIS NCVADYSVLY NSASFSTFKC YGVSPTKLND LCFTNVYADS FVIRGDEVRQ IAPG QTGKI ADYNYKLPDD FTGCVIAWNS NNLDSKVGGN YNYLYRLFRK SNLKPFERDI STEIYQAGST PCNGVEGFNC YFPLQ SYGF QPTNGVGYQP YRVVVLSFEL LHAPATVCGP KKSTNLVKNK CVNFNFNGLT GTGVLTESNK KFLPFQQFGR DIADTT DAV RDPQTLEILD ITPCSFGGVS VITPGTNTSN QVAVLYQDVN CTEVPVAIHA DQLTPTWRVY STGSNVFQTR AGCLIGA EH VNNSYECDIP IGAGICASYQ TQTNSPGSAS SVASQSIIAY TMSLGAENSV AYSNNSIAIP TNFTISVTTE ILPVSMTK T SVDCTMYICG DSTECSNLLL QYGSFCTQLN RALTGIAVEQ DKNTQEVFAQ VKQIYKTPPI KDFGGFNFSQ ILPDPSKPS KRSFIEDLLF NKVTLADAGF IKQYGDCLGD IAARDLICAQ KFNGLTVLPP LLTDEMIAQY TSALLAGTIT SGWTFGAGAA LQIPFAMQM AYRFNGIGVT QNVLYENQKL IANQFNSAIG KIQDSLSSTA SALGKLQDVV NQNAQALNTL VKQLSSNFGA I SSVLNDIL SRLDPPEAEV QIDRLITGRL QSLQTYVTQQ LIRAAEIRAS ANLAATKMSE CVLGQSKRVD FCGKGYHLMS FP QSAPHGV VFLHVTYVPA QEKNFTTAPA ICHDGKAHFP REGVFVSNGT HWFVTQRNFY EPQIITTDNT FVSGNCDVVI GIV NNTVYD PLQPELDSFK EELDKYFKNH TSPDVDLGDI SGINASVVNI QKEIDRLNEV AKNLNESLID LQELGKYEQG SGYI PEAPR DGQAYVRKDG EWVLLSTFLG RSLEVLFQGP GHHHHHHHHS AWSHPQFEKG GGSGGGGSGG SAWSHPQFEK UniProtKB: Spike glycoprotein |
-Macromolecule #3: 2-acetamido-2-deoxy-beta-D-glucopyranose
| Macromolecule | Name: 2-acetamido-2-deoxy-beta-D-glucopyranose / type: ligand / ID: 3 / Number of copies: 26 / Formula: NAG |
|---|---|
| Molecular weight | Theoretical: 221.208 Da |
| Chemical component information |  ChemComp-NAG: |
-Experimental details
-Structure determination
| Method | cryo EM |
|---|---|
 Processing Processing | single particle reconstruction |
| Aggregation state | particle |
-Sample preparation
| Concentration | 0.5 mg/mL |
|---|---|
| Buffer | pH: 5.5 / Component - Concentration: 0.1 M / Component - Name: Acetate |
| Grid | Model: Quantifoil R2/2 / Material: GOLD / Mesh: 200 / Support film - Material: CARBON / Support film - topology: HOLEY ARRAY / Pretreatment - Type: GLOW DISCHARGE / Pretreatment - Time: 30 sec. / Pretreatment - Atmosphere: AIR / Pretreatment - Pressure: 0.039 kPa |
| Vitrification | Cryogen name: ETHANE / Chamber humidity: 95 % / Chamber temperature: 277 K / Instrument: FEI VITROBOT MARK IV |
- Electron microscopy
Electron microscopy
| Microscope | FEI TITAN KRIOS |
|---|---|
| Specialist optics | Energy filter - Name: GIF Bioquantum / Energy filter - Slit width: 20 eV |
| Image recording | Film or detector model: GATAN K3 BIOQUANTUM (6k x 4k) / Digitization - Dimensions - Width: 5760 pixel / Digitization - Dimensions - Height: 4092 pixel / Number grids imaged: 1 / Number real images: 9816 / Average exposure time: 2.18 sec. / Average electron dose: 40.0 e/Å2 |
| Electron beam | Acceleration voltage: 300 kV / Electron source:  FIELD EMISSION GUN FIELD EMISSION GUN |
| Electron optics | C2 aperture diameter: 100.0 µm / Illumination mode: FLOOD BEAM / Imaging mode: BRIGHT FIELD / Cs: 2.7 mm / Nominal defocus max: 2.5 µm / Nominal defocus min: 1.25 µm / Nominal magnification: 105000 |
| Sample stage | Specimen holder model: FEI TITAN KRIOS AUTOGRID HOLDER / Cooling holder cryogen: NITROGEN |
| Experimental equipment |  Model: Titan Krios / Image courtesy: FEI Company |